scCS Tutorial — Single-Condition Analysis¶
Single-cell Commitment Scores with Radial Star Embedding¶
scCS v0.7.0 computes RNA velocity-based commitment scores for k-furcation cell fate decisions.
What scCS does¶
Takes a user-defined bifurcation cluster (progenitor) and k terminal fate clusters
Builds a radial star embedding — progenitor at origin, each fate on its own arm
Projects RNA velocity into this embedding
Computes commitment scores (unCS, nCS) and per-cell fate affinities
Optionally bootstraps confidence intervals on all scores
Identifies driver genes (velocity-based + DEG-based) per fate arm
Runs pathway enrichment (KEGG, GO BP, Reactome) per fate
Reference¶
Kriukov et al. (2025) Single-cell transcriptome of myeloid cells in response to transplantation of human retinal neurons reveals reversibility of microglial activation
Notebook outline
Section |
What you learn |
|---|---|
|
pip install |
|
scVelo pancreas dataset |
|
scVelo dynamical model |
|
|
|
|
|
|
|
star plot, rose, pairwise heatmap, bar chart |
|
fate affinity, entropy, NN-smoothed entropy |
|
write scores back to full adata |
|
|
|
velocity drivers, DEG drivers |
|
KEGG / GO BP per fate |
|
|
1. Installation¶
[1]:
%matplotlib inline
# Install from GitHub (recommended)
# !pip install git+https://github.com/mcrewcow/scCS.git
# Or from a local directory:
#!pip install -e /...
import scCS
print(f"scCS version: {scCS.__version__}")
scCS version: 0.7.4
2. Load & preprocess data¶
We use the built-in pancreas dataset from scVelo — a well-studied k=4 bifurcation (Ductal → Alpha, Beta, Delta, Epsilon). The dataset already contains spliced/unspliced counts and cluster annotations.
[2]:
import numpy as np
import pandas as pd
import anndata
import matplotlib.pyplot as plt
import scanpy as sc
import scvelo as scv
import warnings
warnings.filterwarnings("ignore", category=DeprecationWarning)
warnings.filterwarnings("ignore", category=FutureWarning)
try:
from statsmodels.tools.sm_exceptions import ConvergenceWarning
warnings.filterwarnings("ignore", category=ConvergenceWarning)
except ImportError:
pass
sc.settings.verbosity = 1
scv.settings.verbosity = 1
adata = scv.datasets.pancreas()
print(adata)
print("\nCluster labels:", adata.obs['clusters'].unique().tolist())
# --- Tutorial display helpers (compact tables instead of per-fate loops) ---
pd.set_option("display.max_rows", 20)
pd.set_option("display.max_columns", 15)
pd.set_option("display.width", 160)
def _stack_drivers(driver_dict, n_per_fate=5, cols=None):
"""Concatenate a {fate: DataFrame} dict into one tidy long-form table.
Shows the top ``n_per_fate`` rows per fate; adds a ``fate`` column at the
front. Pass ``cols`` to pick a subset of columns to display.
"""
out = []
for fate, df in driver_dict.items():
if df is None or len(df) == 0:
continue
block = df.head(n_per_fate).copy()
block.insert(0, "fate", fate)
out.append(block)
if not out:
return pd.DataFrame()
big = pd.concat(out, ignore_index=True)
if cols is not None:
keep = ["fate"] + [c for c in cols if c in big.columns]
big = big[keep]
return big
def _stack_enrichment(enrichment_dict, n_per_group=3):
"""Flatten enrichment {fate: {direction: df}} into one tidy table."""
rows = []
for fate, fate_results in enrichment_dict.items():
for direction in ("up", "down"):
df = fate_results.get(direction, None)
if df is None or df.empty:
continue
top = df.head(n_per_group).copy()
top.insert(0, "direction", direction)
top.insert(0, "fate", fate)
rows.append(top)
if not rows:
return pd.DataFrame()
return pd.concat(rows, ignore_index=True)
AnnData object with n_obs × n_vars = 3696 × 27998
obs: 'clusters_coarse', 'clusters', 'S_score', 'G2M_score'
var: 'highly_variable_genes'
uns: 'clusters_coarse_colors', 'clusters_colors', 'day_colors', 'neighbors', 'pca'
obsm: 'X_pca', 'X_umap'
layers: 'spliced', 'unspliced'
obsp: 'distances', 'connectivities'
Cluster labels: ['Pre-endocrine', 'Ductal', 'Alpha', 'Ngn3 high EP', 'Delta', 'Beta', 'Ngn3 low EP', 'Epsilon']
[3]:
# Standard scVelo preprocessing
import inspect as _inspect
_fn_sig = _inspect.signature(scv.pp.filter_and_normalize).parameters
if "n_top_genes" in _fn_sig:
scv.pp.filter_and_normalize(adata, min_shared_counts=20, n_top_genes=2000)
else:
# scvelo builds without n_top_genes: do filter_and_normalize then HVG.
# Use flavor="cell_ranger" not the default "seurat": seurat passes
# int n_bins to pd.cut on log-dispersions which contain +/-inf for
# low-mean genes — pandas >= 2.2 rejects this with ValueError.
# cell_ranger uses explicit bin edges including +/-inf and is
# robust across pandas/py versions (scCS changelog v0.7.4).
scv.pp.filter_and_normalize(adata, min_shared_counts=20)
sc.pp.highly_variable_genes(
adata, n_top_genes=2000, subset=True, flavor="cell_ranger"
)
sc.pp.neighbors(adata)
scv.pp.moments(adata, n_pcs=None, n_neighbors=None)
3. RNA velocity¶
We run the dynamical model (most accurate) via scVelo. If your data already has velocity and velocity_graph, skip this section.
[4]:
scv.tl.recover_dynamics(adata, n_jobs=32)
scv.tl.velocity(adata, mode="dynamical")
scv.tl.velocity_graph(adata)
scv.tl.velocity_pseudotime(adata)
# Visualise velocity on UMAP
scv.pl.velocity_embedding_stream(adata, basis="umap", color="clusters",
title="RNA velocity — pancreas")
/home/emil/miniforge3/envs/lab-py312/lib/python3.12/multiprocessing/popen_fork.py:66: DeprecationWarning: This process (pid=3974) is multi-threaded, use of fork() may lead to deadlocks in the child.
self.pid = os.fork()
/home/emil/miniforge3/envs/lab-py312/lib/python3.12/multiprocessing/popen_fork.py:66: DeprecationWarning: This process (pid=3974) is multi-threaded, use of fork() may lead to deadlocks in the child.
self.pid = os.fork()
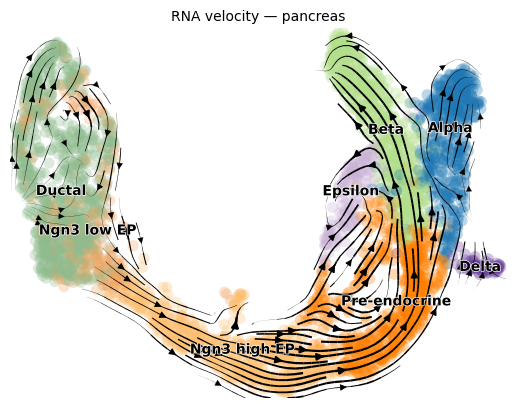
Alternatively, you can let scCS run the full velocity pipeline for you:
scorer = scCS.SingleScorer(adata, ...)
scorer.compute_velocity(mode="dynamical", n_top_genes=2000)
4. Build the radial star embedding¶
The pancreas dataset has a clear bifurcation from Ductal progenitors into four terminal fates: Alpha, Beta, Delta, Epsilon.
SingleScorer requires:
root— the progenitor cluster labelbranches— list of terminal fate cluster labelsobs_key— column inadata.obswith cluster labels
[5]:
scorer = scCS.SingleScorer(
adata,
root="Pre-endocrine",
branches=["Alpha", "Beta", "Delta", "Epsilon"],
obs_key="clusters",
n_angle_bins=36, # 10° angular bins (default)
sector_method="centroid", # anchor sectors to fate centroids (recommended)
)
# Build the star embedding using scVelo pseudotime
scorer.build_embedding(
ordering_metric="pseudotime", # uses adata.obs['velocity_pseudotime']
arm_scale=10.0, # maximum radial distance
jitter=0.3, # perpendicular noise to avoid overplotting
)
print("Embedding shape:", scorer.adata_sub.obsm["X_sccs"].shape)
print("Cells in embedding:", scorer.adata_sub.n_obs)
[scCS] Building star embedding: root='Pre-endocrine', k=4 fates, metric='pseudotime'
[scCS] Subsetting: 1876 / 3696 cells kept
(1820 cells from other populations excluded)
Alpha: 481 cells (fate)
Beta: 591 cells (fate)
Delta: 70 cells (fate)
Epsilon: 142 cells (fate)
Pre-endocrine: 592 cells (progenitor)
[scCS] Star embedding built → adata_sub.obsm["X_sccs"] shape: (1876, 2)
Arm angles: {'Alpha': 0.0, 'Beta': 90.0, 'Delta': 180.0, 'Epsilon': 270.0}
[scCS] Star embedding stored in scorer.adata_sub.obsm['X_sccs']. (1876 cells)
Embedding shape: (1876, 2)
Cells in embedding: 1876
5. Fix arm coverage with subset-local pseudotime¶
Problem: scVelo pseudotime is computed on the full adata. When we subset to bifurcation + terminal fate cells, the pseudotime range is compressed — cells cluster near the origin instead of spanning the full arm length.
Solution: refit_pseudotime() extracts the velocity_graph submatrix for the subset cells, recomputes pseudotime locally, scales it to [0, 1], and rebuilds the embedding.
When to use ``scale_01=True`` vs ``False``:
scale_01=True(default) — cells span the full arm; best for single-condition analysis
scale_01=False— preserves absolute pseudotime ordering; use for multi-condition comparisons where cross-condition pseudotime comparability matters
[6]:
# Recompute pseudotime on the subset subgraph and rebuild the embedding
scorer.refit_pseudotime(scale_01=True)
# Inspect the corrected pseudotime
pt_sub = scorer.adata_sub.obs["velocity_pseudotime"]
print(f"Subset pseudotime range: [{pt_sub.min():.3f}, {pt_sub.max():.3f}]")
[scCS] Recomputing pseudotime on subset (1876 / 3696 cells)...
[scCS] Used scanpy DPT (with diffmap) as pseudotime fallback.
[scCS] Subset pseudotime scaled to [0, 1].
[scCS] Subset pseudotime stored in adata_sub.obs['sccs_pseudotime']. Range: [0.000, 1.000]
[scCS] Rebuilding star embedding with subset-local pseudotime...
[scCS] Subsetting: 1876 / 3696 cells kept
(1820 cells from other populations excluded)
Alpha: 481 cells (fate)
Beta: 591 cells (fate)
Delta: 70 cells (fate)
Epsilon: 142 cells (fate)
Pre-endocrine: 592 cells (progenitor)
[scCS] Star embedding built → adata_sub.obsm["X_sccs"] shape: (1876, 2)
Arm angles: {'Alpha': 0.0, 'Beta': 90.0, 'Delta': 180.0, 'Epsilon': 270.0}
[scCS] Embedding rebuilt. Call fit() again to update the FateMap and velocity projection.
Subset pseudotime range: [0.495, 1.000]
6. Fit and compute commitment scores¶
fit() builds the FateMap (validates clusters, computes fate centroids, projects velocity). score() computes all commitment scores.
Key outputs in CommitmentScoreResult¶
Attribute |
Description |
|---|---|
|
Unnormalized CS matrix (k × k) |
|
Cell-count-normalized CS matrix (k × k) |
|
Fraction of velocity mass per fate |
|
Population-level commitment entropy |
|
Mean per-cell fate entropy |
|
Per-fate binary entropy |
|
Per-cell fate affinity matrix (n_cells × k) |
|
Bootstrap CI dict (if |
[7]:
scorer.fit()
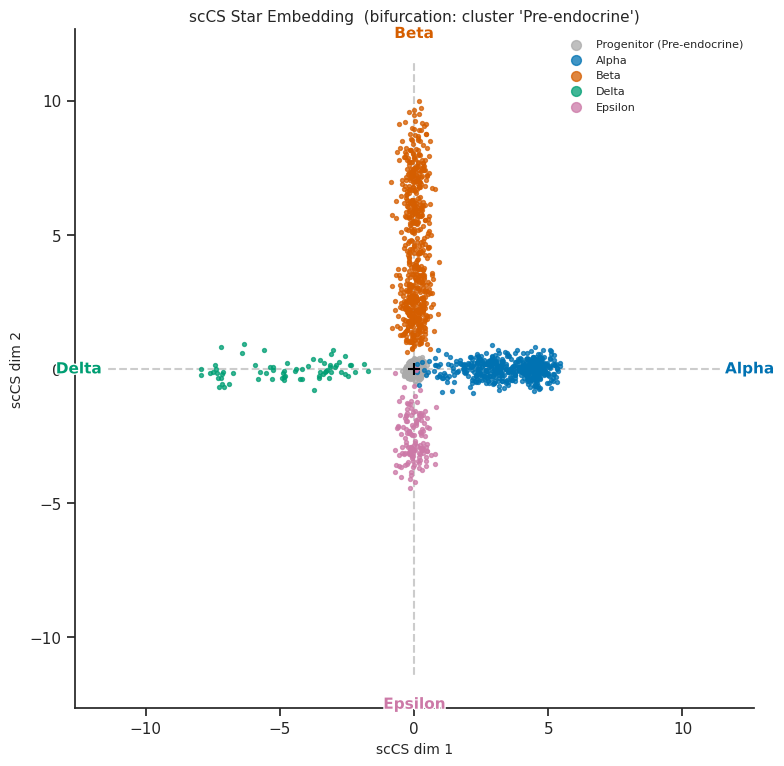
[scCS] Root cluster 'Pre-endocrine': 592 cells, centroid=(-0.00, -0.00)
[scCS] Fate 'Alpha': 481 cells, centroid=(3.51, -0.01)
[scCS] Fate 'Beta': 591 cells, centroid=(0.03, 4.38)
[scCS] Fate 'Delta': 70 cells, centroid=(-4.75, -0.05)
[scCS] Fate 'Epsilon': 142 cells, centroid=(-0.02, -2.55)
[scCS] FateMap built: k=4 fates
[scCS] Projecting velocity via scVelo on full adata → slicing to subset...
[scCS] Velocity projected. Shape: (1876, 2)
FateMap (root='Pre-endocrine', k=4)
Cluster key : 'clusters'
Root cells : 592
Root centroid: (-0.005, -0.003)
Fate 0: 'Alpha' n_cells=481 centroid=(3.51, -0.01) arm_angle=0.0°
Fate 1: 'Beta' n_cells=591 centroid=(0.03, 4.38) arm_angle=90.0°
Fate 2: 'Delta' n_cells=70 centroid=(-4.75, -0.05) arm_angle=180.0°
Fate 3: 'Epsilon' n_cells=142 centroid=(-0.02, -2.55) arm_angle=270.0°
[7]:
SingleScorer(root='Pre-endocrine', branches=['Alpha', 'Beta', 'Delta', 'Epsilon'], k=4, status='fitted')
[8]:
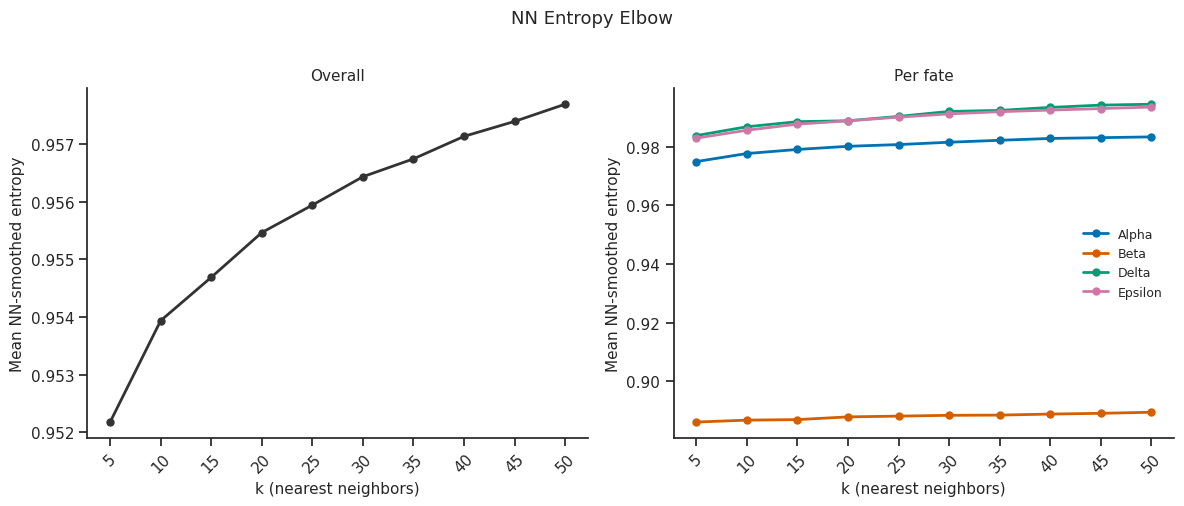
fig = scorer.plot_nn_entropy_elbow(k_nn_range=range(5, 51, 5))
plt.show()
[9]:
# Score with bootstrap confidence intervals (n_bootstrap=500 recommended)
result = scorer.score(
cell_level=True, # compute per-cell fate affinities
k_nn=15, # NN-smoothed entropy (15 nearest neighbours)
n_bootstrap=500, # bootstrap CI on nCS
bootstrap_ci=0.95, # 95% CI
)
[scCS] Computing bootstrap CI (n=500)...
=== CommitmentScoreResult ===
Fates (4): Alpha, Beta, Delta, Epsilon
Dominant fate: Beta
Entropy metrics:
Population entropy: 0.7052 [aggregate velocity-mass balance]
Mean cell entropy: 0.9361 [per-cell average, k-way]
Per-fate cell entropy:
Alpha: 0.8246
Beta: 0.8608
Delta: 0.7541
Epsilon: 0.6399
NN-smoothed entropy (k=15): mean=0.9547 [per-cell, stored in adata_sub.obs['cs_nn_entropy']]
Commitment vector (normalized):
Alpha: 0.2210
Beta: 0.6449
Delta: 0.0765
Epsilon: 0.0576
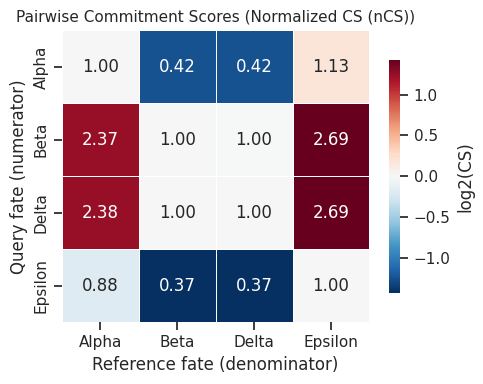
Pairwise nCS matrix:
Alpha Beta Delta Epsilon
Alpha 1.000000 0.421157 0.420532 1.133082
Beta 2.374413 1.000000 0.998516 2.690405
Delta 2.377942 1.001486 1.000000 2.694404
Epsilon 0.882548 0.371691 0.371140 1.000000
Bootstrap 95% CI on nCS (n=500):
CI low:
Alpha Beta Delta Epsilon
Alpha 1.000 0.369 0.352 0.922
Beta 2.119 1.000 0.851 2.270
Delta 1.989 0.855 1.000 2.103
Epsilon 0.723 0.303 0.290 1.000
CI high:
Alpha Beta Delta Epsilon
Alpha 1.000 0.472 0.503 1.383
Beta 2.712 1.000 1.170 3.297
Delta 2.838 1.175 1.000 3.449
Epsilon 1.084 0.440 0.476 1.000
[10]:
# Summary table
print(result.summary())
=== CommitmentScoreResult ===
Fates (4): Alpha, Beta, Delta, Epsilon
Dominant fate: Beta
Entropy metrics:
Population entropy: 0.7052 [aggregate velocity-mass balance]
Mean cell entropy: 0.9361 [per-cell average, k-way]
Per-fate cell entropy:
Alpha: 0.8246
Beta: 0.8608
Delta: 0.7541
Epsilon: 0.6399
NN-smoothed entropy (k=15): mean=0.9547 [per-cell, stored in adata_sub.obs['cs_nn_entropy']]
Commitment vector (normalized):
Alpha: 0.2210
Beta: 0.6449
Delta: 0.0765
Epsilon: 0.0576
Pairwise nCS matrix:
Alpha Beta Delta Epsilon
Alpha 1.000000 0.421157 0.420532 1.133082
Beta 2.374413 1.000000 0.998516 2.690405
Delta 2.377942 1.001486 1.000000 2.694404
Epsilon 0.882548 0.371691 0.371140 1.000000
Bootstrap 95% CI on nCS (n=500):
CI low:
Alpha Beta Delta Epsilon
Alpha 1.000 0.369 0.352 0.922
Beta 2.119 1.000 0.851 2.270
Delta 1.989 0.855 1.000 2.103
Epsilon 0.723 0.303 0.290 1.000
CI high:
Alpha Beta Delta Epsilon
Alpha 1.000 0.472 0.503 1.383
Beta 2.712 1.000 1.170 3.297
Delta 2.838 1.175 1.000 3.449
Epsilon 1.084 0.440 0.476 1.000
[11]:
# Access individual scores
print("Pairwise nCS matrix:")
print(pd.DataFrame(
result.pairwise_nCS,
index=result.fate_names,
columns=result.fate_names
).round(3))
print("\nCommitment vector (fraction of velocity mass per fate):")
for fate, cv in zip(result.fate_names, result.commitment_vector):
print(f" {fate}: {cv:.3f}")
print(f"\nPopulation entropy: {result.population_entropy:.3f}")
print(f"Mean per-cell entropy: {result.mean_cell_entropy:.3f}")
Pairwise nCS matrix:
Alpha Beta Delta Epsilon
Alpha 1.000 0.421 0.421 1.133
Beta 2.374 1.000 0.999 2.690
Delta 2.378 1.001 1.000 2.694
Epsilon 0.883 0.372 0.371 1.000
Commitment vector (fraction of velocity mass per fate):
Alpha: 0.221
Beta: 0.645
Delta: 0.076
Epsilon: 0.058
Population entropy: 0.705
Mean per-cell entropy: 0.936
[12]:
# Bootstrap CI (if n_bootstrap > 0)
if result.bootstrap_ci is not None:
ci = result.bootstrap_ci
print(f"Bootstrap CI ({int(ci['ci_level']*100)}%, n={ci['n_bootstrap']}):")
print(" nCS CI low:")
print(pd.DataFrame(ci['ci_low'], index=result.fate_names, columns=result.fate_names).round(3))
print("\n nCS CI high:")
print(pd.DataFrame(ci['ci_high'], index=result.fate_names, columns=result.fate_names).round(3))
Bootstrap CI (95%, n=500):
nCS CI low:
Alpha Beta Delta Epsilon
Alpha 1.000 0.369 0.352 0.922
Beta 2.119 1.000 0.851 2.270
Delta 1.989 0.855 1.000 2.103
Epsilon 0.723 0.303 0.290 1.000
nCS CI high:
Alpha Beta Delta Epsilon
Alpha 1.000 0.472 0.503 1.383
Beta 2.712 1.000 1.170 3.297
Delta 2.838 1.175 1.000 3.449
Epsilon 1.084 0.440 0.476 1.000
7. Visualizations¶
scCS provides several built-in plots. All are accessible as methods on the scorer or as standalone functions in scCS.plot.
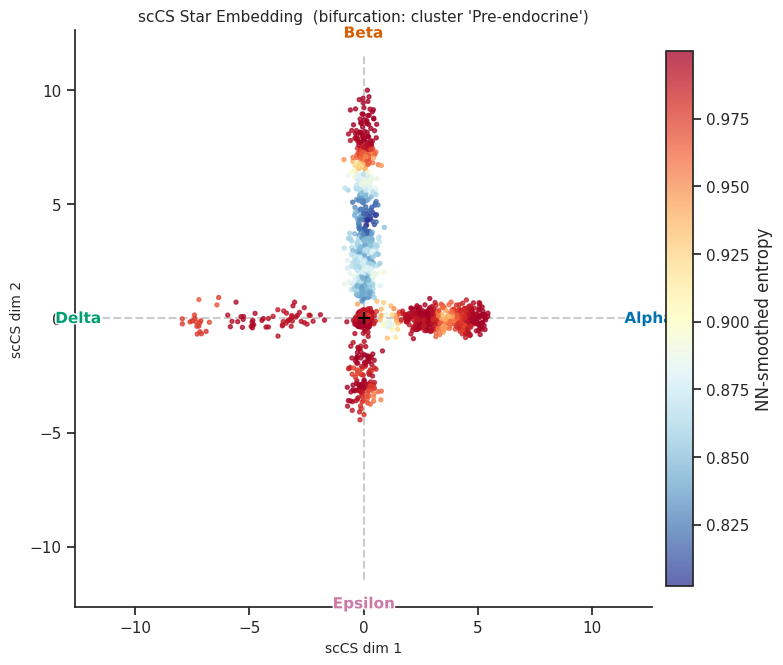
7.1 Radial star embedding¶
[13]:
# Primary visualization: cells in the star embedding, coloured by fate
fig = scorer.plot_star(result, figsize=(8, 8))
plt.tight_layout()
plt.show()
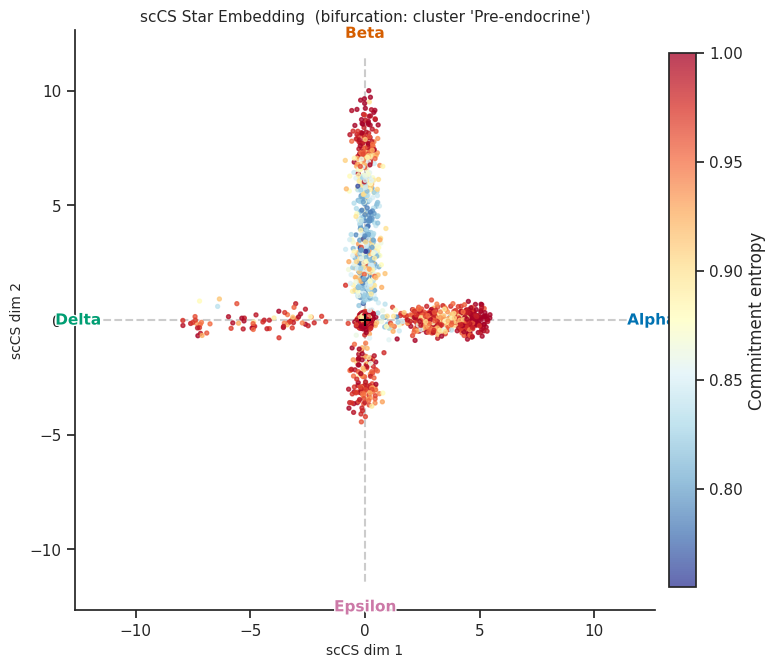
[14]:
# Colour by per-cell entropy instead of fate
fig = scorer.plot_star(result, color_by="entropy", figsize=(8, 8))
plt.tight_layout()
plt.show()
[15]:
# Colour by NN-smoothed entropy (requires k_nn in score())
fig = scorer.plot_star(result, color_by="nn_entropy", figsize=(8, 8))
plt.tight_layout()
plt.show()
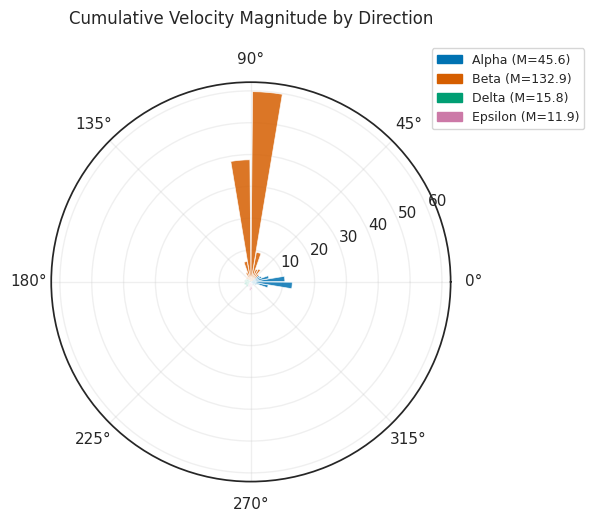
7.2 Rose / polar plot¶
[16]:
# Rose plot: cumulative velocity magnitude per angular bin
fig = scorer.plot_rose(result, figsize=(6, 6))
plt.tight_layout()
plt.show()
7.3 Pairwise nCS heatmap¶
[17]:
# Heatmap of pairwise normalized commitment scores
fig = scorer.plot_pairwise_cs(result, figsize=(5, 4))
plt.tight_layout()
plt.show()
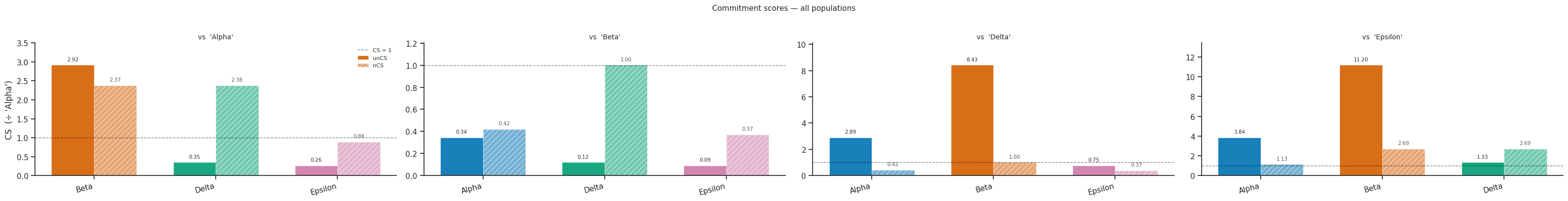
7.4 Commitment bar chart¶
[18]:
# Bar chart: unCS vs nCS per fate pair
fig = scorer.plot_commitment_bar(result, figsize=(8, 4))
plt.tight_layout()
plt.show()
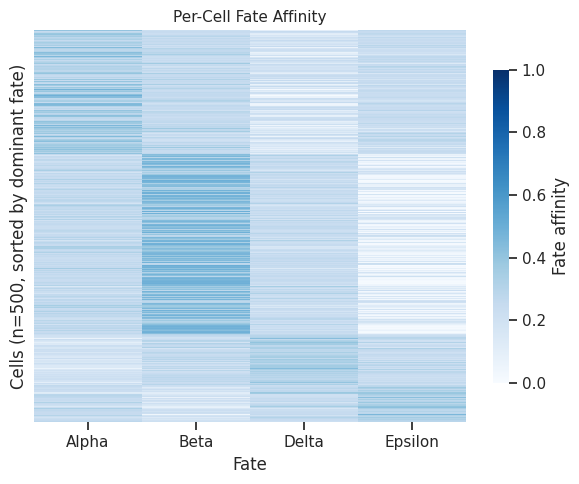
7.5 Per-cell fate affinity heatmap¶
[19]:
# Heatmap of per-cell fate affinities (rows = cells, cols = fates)
fig = scorer.plot_commitment_heatmap(result, figsize=(6, 5), max_cells=500)
plt.tight_layout()
plt.show()
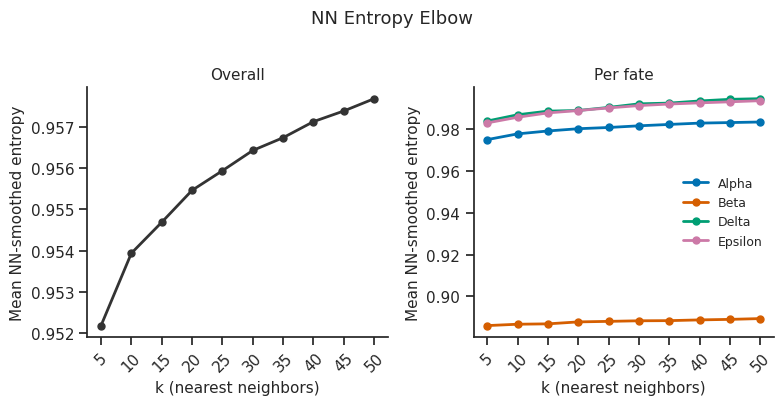
7.6 NN entropy elbow plot¶
[20]:
# Elbow plot to choose k_nn for NN-smoothed entropy
# Sweeps k_nn values and plots mean NN entropy
fig = scorer.plot_nn_entropy_elbow(k_nn_range=range(5, 51, 5), figsize=(8, 4))
plt.tight_layout()
plt.show()
8. Per-cell fate affinity scores¶
After score(cell_level=True), per-cell scores are stored in scorer.adata_sub.obs:
Column |
Description |
|---|---|
|
Per-cell affinity for each fate (sums to 1 per cell) |
|
Fate with highest affinity |
|
Per-cell commitment entropy (0 = fully committed, 1 = uniform) |
|
NN-smoothed entropy (if |
|
Subset-local pseudotime |
[21]:
# Inspect per-cell scores
obs_cols = [c for c in scorer.adata_sub.obs.columns if c.startswith("cs_")]
print("Per-cell score columns:", obs_cols)
scorer.adata_sub.obs[obs_cols].head(10)
Per-cell score columns: ['cs_Alpha', 'cs_Beta', 'cs_Delta', 'cs_Epsilon', 'cs_dominant_fate', 'cs_entropy', 'cs_nn_entropy']
[21]:
| cs_Alpha | cs_Beta | cs_Delta | cs_Epsilon | cs_dominant_fate | cs_entropy | cs_nn_entropy | |
|---|---|---|---|---|---|---|---|
| index | |||||||
| AAACCTGAGAGGGATA | 0.345656 | 0.252071 | 0.154299 | 0.247973 | Alpha | 0.972886 | 0.992053 |
| AAACCTGAGGCAATTA | 0.247055 | 0.296687 | 0.252668 | 0.203590 | Beta | 0.993692 | 0.992740 |
| AAACCTGTCCCTCTTT | 0.324502 | 0.244887 | 0.175503 | 0.255107 | Alpha | 0.983665 | 0.958981 |
| AAACGGGAGTAGCGGT | 0.361040 | 0.324278 | 0.138481 | 0.176201 | Alpha | 0.946906 | 0.977282 |
| AAACGGGCAAAGAATC | 0.274640 | 0.476842 | 0.224002 | 0.024516 | Beta | 0.818077 | 0.875318 |
| AAACGGGGTACAGTTC | 0.251495 | 0.463168 | 0.247236 | 0.038101 | Beta | 0.846585 | 0.884746 |
| AAACGGGGTCGGGTCT | 0.268075 | 0.221469 | 0.232088 | 0.278367 | Epsilon | 0.996724 | 0.998418 |
| AAACGGGTCCGCGCAA | 0.328827 | 0.381496 | 0.170364 | 0.119313 | Beta | 0.929480 | 0.988123 |
| AAACGGGTCGCATGGC | 0.272847 | 0.479499 | 0.225780 | 0.021874 | Beta | 0.812556 | 0.817800 |
| AAAGATGAGAGTACCG | 0.213046 | 0.270671 | 0.286844 | 0.229439 | Delta | 0.994829 | 0.992597 |
[22]:
# Distribution of dominant fate assignments
print(scorer.adata_sub.obs["cs_dominant_fate"].value_counts())
cs_dominant_fate
Beta 836
Alpha 604
Delta 246
Epsilon 190
Name: count, dtype: int64
[23]:
# Entropy distribution per fate
import matplotlib.pyplot as plt
fig, axes = plt.subplots(1, len(result.fate_names), figsize=(12, 3), sharey=True)
for ax, fate in zip(axes, result.fate_names):
mask = scorer.adata_sub.obs["cs_dominant_fate"] == fate
ax.hist(scorer.adata_sub.obs.loc[mask, "cs_entropy"], bins=20, color="steelblue", edgecolor="white")
ax.set_title(fate)
ax.set_xlabel("Entropy")
axes[0].set_ylabel("# cells")
plt.suptitle("Per-cell commitment entropy by dominant fate", y=1.02)
plt.tight_layout()
plt.show()
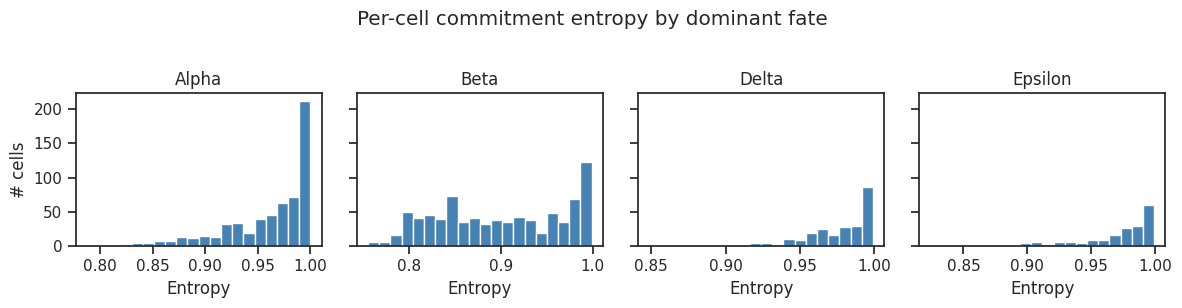
9. Transfer labels to full adata¶
transfer_labels() writes per-cell scores from adata_sub back to the full adata, so you can use them in UMAP plots, downstream analyses, or integration with other tools.
Cells not in the embedding subset receive NaN (numeric) or 'unassigned' (categorical).
[24]:
scorer.transfer_labels(adata, result, prefix="cs_")
# Columns added to adata.obs
cs_cols = [c for c in adata.obs.columns if c.startswith("cs_")]
print("Columns added to adata.obs:", cs_cols)
adata.obs[cs_cols].head()
[scCS] Labels transferred to adata.obs for 1876 / 3696 cells. Columns: ['cs_Alpha', 'cs_Beta', 'cs_Delta', 'cs_Epsilon', 'cs_dominant_fate', 'cs_entropy']
Columns added to adata.obs: ['cs_Alpha', 'cs_Beta', 'cs_Delta', 'cs_Epsilon', 'cs_dominant_fate', 'cs_entropy', 'cs_nn_entropy']
[24]:
| cs_Alpha | cs_Beta | cs_Delta | cs_Epsilon | cs_dominant_fate | cs_entropy | cs_nn_entropy | |
|---|---|---|---|---|---|---|---|
| index | |||||||
| AAACCTGAGAGGGATA | 0.345656 | 0.252071 | 0.154299 | 0.247973 | Alpha | 0.972886 | 0.992053 |
| AAACCTGAGCCTTGAT | NaN | NaN | NaN | NaN | unassigned | NaN | NaN |
| AAACCTGAGGCAATTA | 0.247055 | 0.296687 | 0.252668 | 0.203590 | Beta | 0.993692 | 0.992740 |
| AAACCTGCATCATCCC | NaN | NaN | NaN | NaN | unassigned | NaN | NaN |
| AAACCTGGTAAGTGGC | NaN | NaN | NaN | NaN | unassigned | NaN | NaN |
[25]:
# Visualize on UMAP
sc.pl.umap(adata, color=["cs_dominant_fate", "cs_entropy", "cs_Alpha", "cs_Beta"],
ncols=2, wspace=0.4)
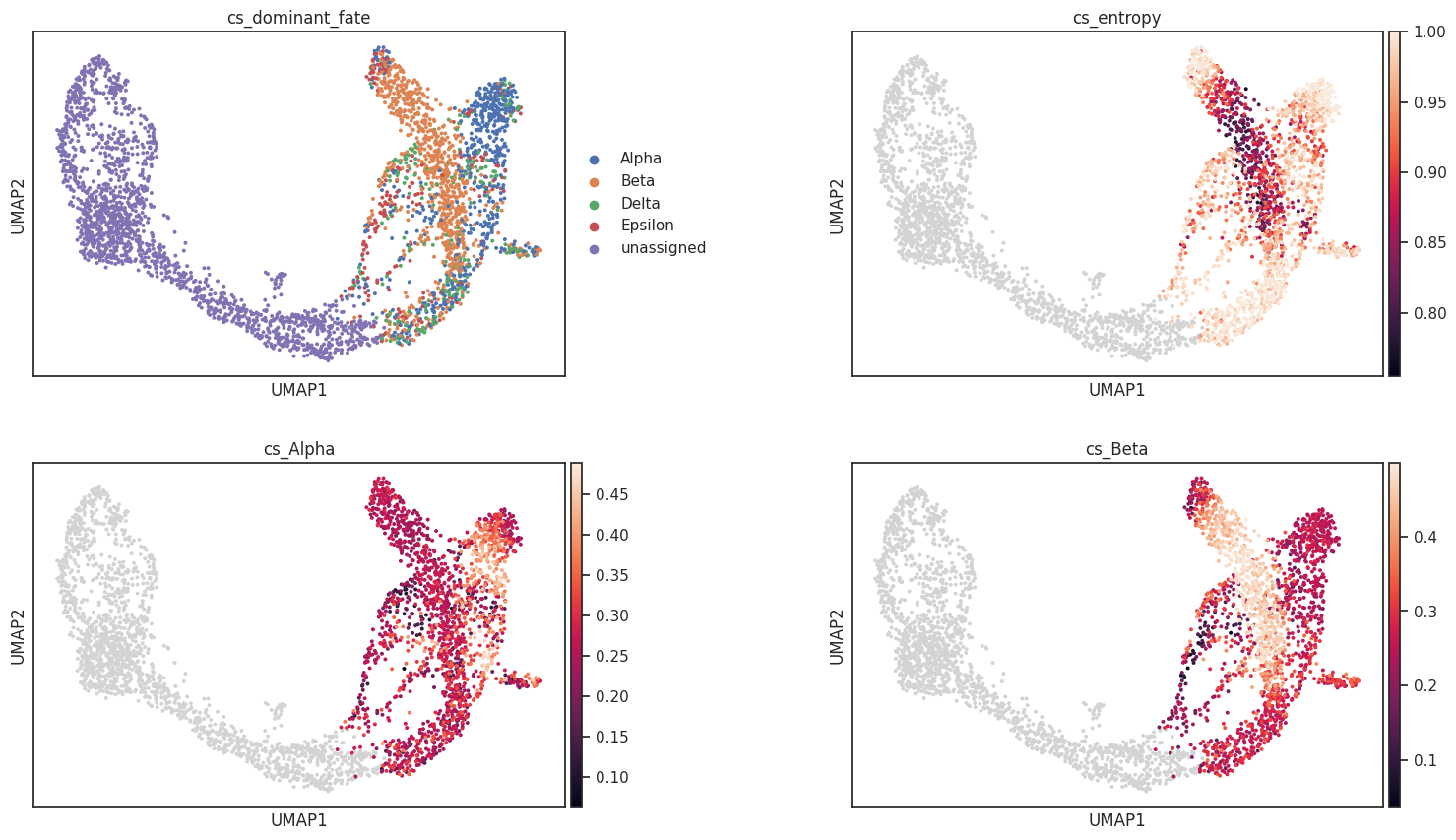
10. Subset scoring¶
score_per_subset() computes commitment scores separately for each value of a metadata column in adata_sub.obs. Useful for comparing commitment across time points, batches, or any categorical variable.
Note: The mask is applied to
adata_sub(the embedding subset), not the full adata.
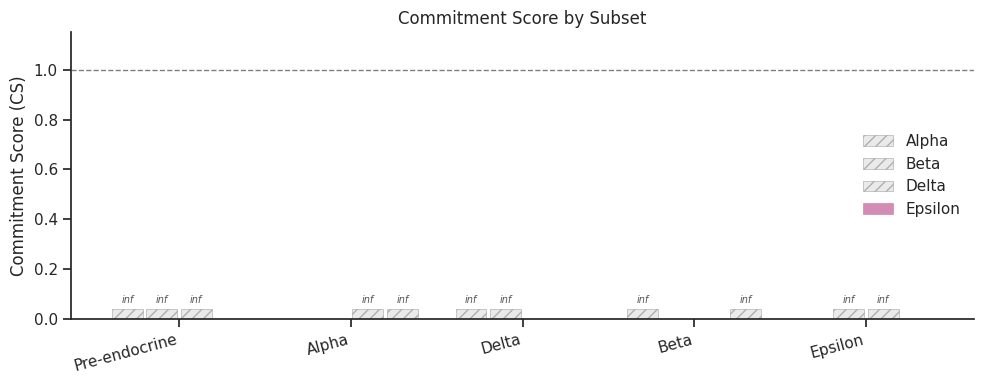
Note on ``inf`` in pairwise nCS: When a subset contains only progenitor cells (e.g., ‘Pre-endocrine’) with no cells from any fate arm,
pairwise_nCSwill beinffor all off-diagonal entries. This is mathematically correct — nCS is undefined when a fate arm has 0 cells. AUserWarningis emitted automatically. Progenitor-only subsets are still useful for inspecting the commitment vector and population entropy.
[26]:
# Score separately for each cluster within the embedding
# (Here we use 'clusters' as a demo; in practice use a condition/batch column)
subset_results = scorer.score_per_subset(
split_by="clusters",
cell_level=False,
n_bootstrap=0,
verbose=False,
)
# Tidy summary: one row per subset with all off-diagonal nCS values + status
def _subset_summary(subset_results):
rows = []
for subset_name, res in subset_results.items():
n = len(res.fate_names)
ncs = res.pairwise_nCS
row = {"subset": subset_name, "n_cells_in_fate": int(res.n_cells_per_fate.sum())}
for i in range(n):
for j in range(i + 1, n):
col = f"nCS[{res.fate_names[i]} vs {res.fate_names[j]}]"
row[col] = float(ncs[i, j])
# Status flag: any inf?
off_diag = [ncs[i, j] for i in range(n) for j in range(n) if i != j]
if all(np.isinf(v) for v in off_diag):
row["status"] = "progenitor-only (inf)"
elif any(np.isinf(v) for v in off_diag):
row["status"] = "partial (some inf)"
else:
row["status"] = "ok"
rows.append(row)
return pd.DataFrame(rows)
summary_df = _subset_summary(subset_results)
summary_df
[26]:
| subset | n_cells_in_fate | nCS[Alpha vs Beta] | nCS[Alpha vs Delta] | nCS[Alpha vs Epsilon] | nCS[Beta vs Delta] | nCS[Beta vs Epsilon] | nCS[Delta vs Epsilon] | status | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | Pre-endocrine | 0 | inf | inf | inf | inf | inf | inf | progenitor-only (inf) |
| 1 | Alpha | 481 | 0.0 | 0.0 | 0.0 | inf | inf | inf | partial (some inf) |
| 2 | Delta | 70 | inf | inf | inf | inf | inf | 0.0 | partial (some inf) |
| 3 | Beta | 591 | inf | inf | inf | 0.0 | 0.0 | inf | partial (some inf) |
| 4 | Epsilon | 142 | inf | inf | inf | inf | inf | inf | partial (some inf) |
[27]:
# Visualize subset comparison
fig = scorer.plot_subset_comparison(subset_results, figsize=(10, 4))
plt.tight_layout()
plt.show()
11. Driver genes¶
scCS provides three complementary approaches to identify fate-driving genes:
Velocity drivers (
get_velocity_drivers) — genes with highest mean RNA velocity in each fate arm (high positive velocity = gene is being upregulated along that fate)DEG drivers — differentially expressed genes between each fate arm and the bifurcation cluster (Wilcoxon rank-sum test)
Velocity-fate correlation drivers (
get_velocity_fate_drivers) — Spearman correlation between each gene’s velocity and per-cell fate affinity. Stronger signal than mean velocity because it filters out genes that are fast everywhere.
[28]:
# Velocity-based driver genes (top 20 per fate)
vel_drivers = scorer.get_velocity_drivers(n_top_genes=20)
# Compact long-form table: top 5 per fate, one DataFrame
print("Top velocity drivers (top 5 per fate):")
_stack_drivers(vel_drivers, n_per_fate=5, cols=["gene", "mean_velocity", "rank"])
── Velocity drivers: Alpha (top 20, sorted by delta_velocity) ──
rank gene delta_velocity mean_velocity
1 Clu 1.488299 1.532060
2 Cpa2 1.242302 0.851096
3 Cryba2 1.057625 1.217162
4 Ghrl 0.986426 0.384223
5 Meis2 0.821635 0.665699
6 Pax6 0.726139 0.883325
7 Npepl1 0.725493 0.182687
8 Tm4sf4 0.668253 0.466062
9 Vasp 0.611339 0.097546
10 Pax4 0.603224 0.034867
11 Btbd17 0.577881 0.930656
12 Krt7 0.547158 -0.280244
13 Cd200 0.535196 0.330072
14 Celf3 0.534336 0.370460
15 Tox3 0.497146 0.251629
16 Pdx1 0.496890 0.392475
17 Ppp3ca 0.487356 0.200745
18 Hn1 0.468364 0.031799
19 BC023829 0.455840 0.336861
20 Cpt2 0.432719 0.246379
── Velocity drivers: Beta (top 20, sorted by delta_velocity) ──
rank gene delta_velocity mean_velocity
1 Ins2 3.672153 3.689225
2 Nnat 3.631306 4.051527
3 Isl1 3.562601 1.070217
4 Clu 1.605093 1.648854
5 1700086L19Rik 1.411600 0.453118
6 Krt7 1.125878 0.298475
7 Pax4 0.901425 0.333068
8 Ppp1r1a 0.861976 1.539815
9 Tm4sf4 0.842035 0.639844
10 Igfbpl1 0.771356 0.268164
11 Cpa2 0.714384 0.323178
12 Pax6 0.697922 0.855108
13 Dbn1 0.615245 0.314196
14 Pdx1 0.535203 0.430788
15 Meis2 0.518604 0.362668
16 BC023829 0.514133 0.395154
17 Cldn6 0.508870 -0.301879
18 Glud1 0.481521 0.737995
19 Smarcd2 0.460625 0.598269
20 Lurap1l 0.456940 0.285098
── Velocity drivers: Delta (top 20, sorted by delta_velocity) ──
rank gene delta_velocity mean_velocity
1 Cpa2 2.483908 2.092702
2 Krt8 2.349361 1.407045
3 Maged2 2.340670 3.380898
4 Cryba2 1.744062 1.903599
5 Ldha 1.710826 1.581080
6 Hn1 1.123444 0.686879
7 Cdkn1c 1.072009 1.007896
8 Nnat 1.059840 1.480061
9 Ppp1r1a 1.017116 1.694955
10 Ambp 0.908994 0.752210
11 Gnas 0.861592 6.604826
12 Pax6 0.851152 1.008338
13 Akr1c19 0.804932 1.123999
14 Sparc 0.687036 0.649847
15 Meis2 0.684853 0.528917
16 Rpl12 0.670421 -0.154604
17 Celf3 0.637237 0.473361
18 BC023829 0.635596 0.516617
19 Krt18 0.603725 -0.206350
20 Foxa3 0.588107 0.411777
── Velocity drivers: Epsilon (top 20, sorted by delta_velocity) ──
rank gene delta_velocity mean_velocity
1 Cpa2 1.345101 0.953895
2 Cryba2 1.310028 1.469565
3 Clu 1.293649 1.337410
4 Meis2 1.209729 1.053792
5 Pax4 0.843907 0.275551
6 Syt13 0.799221 0.825314
7 Siva1 0.731447 0.615843
8 Pax6 0.731163 0.888349
9 Lurap1l 0.654209 0.482367
10 Arl6ip1 0.602216 -0.050500
11 Nudt19 0.557069 0.466408
12 Npepl1 0.499464 -0.043343
13 Pdx1 0.488036 0.383621
14 Rgs17 0.487121 0.559517
15 Gars 0.450410 0.620145
16 Cpt2 0.450140 0.263800
17 Cdkn1c 0.401330 0.337217
18 Ppp1r14b 0.393609 -0.294763
19 Cdk1 0.393504 0.399177
20 Tkt 0.385895 0.397531
Top velocity drivers (top 5 per fate):
[28]:
| fate | gene | mean_velocity | rank | |
|---|---|---|---|---|
| 0 | Alpha | Clu | 1.532060 | 1 |
| 1 | Alpha | Cpa2 | 0.851096 | 2 |
| 2 | Alpha | Cryba2 | 1.217162 | 3 |
| 3 | Alpha | Ghrl | 0.384223 | 4 |
| 4 | Alpha | Meis2 | 0.665699 | 5 |
| 5 | Beta | Ins2 | 3.689225 | 1 |
| 6 | Beta | Nnat | 4.051527 | 2 |
| 7 | Beta | Isl1 | 1.070217 | 3 |
| 8 | Beta | Clu | 1.648854 | 4 |
| 9 | Beta | 1700086L19Rik | 0.453118 | 5 |
| 10 | Delta | Cpa2 | 2.092702 | 1 |
| 11 | Delta | Krt8 | 1.407045 | 2 |
| 12 | Delta | Maged2 | 3.380898 | 3 |
| 13 | Delta | Cryba2 | 1.903599 | 4 |
| 14 | Delta | Ldha | 1.581080 | 5 |
| 15 | Epsilon | Cpa2 | 0.953895 | 1 |
| 16 | Epsilon | Cryba2 | 1.469565 | 2 |
| 17 | Epsilon | Clu | 1.337410 | 3 |
| 18 | Epsilon | Meis2 | 1.053792 | 4 |
| 19 | Epsilon | Pax4 | 0.275551 | 5 |
[29]:
# DEG-based driver genes (top 20 per fate, vs the bifurcation cluster)
deg_drivers = scorer.get_deg_drivers(
n_top_genes=20,
pval_threshold=0.05,
logfc_threshold=0.25,
)
print("Top DEG drivers (top 5 per fate):")
_stack_drivers(
deg_drivers, n_per_fate=5,
cols=["gene", "logfoldchange", "pval_adj", "pct_fate", "pct_progenitor"],
)
── DEG drivers: Alpha vs progenitor ──
Significant: 761 (up: 376, down: 385)
gene logfoldchange pval_adj pct_fate pct_progenitor
Pyy 209.768677 1.181761e-93 0.968815 NaN
Gcg 209.614731 9.196179e-64 0.656965 NaN
Iapp 99.370232 4.065913e-99 0.854470 NaN
Ttr 67.718864 2.617159e-103 1.000000 NaN
Gnas 34.805965 2.937512e-52 1.000000 NaN
Rbp4 33.815922 7.031753e-50 0.962578 NaN
Hpgds 26.723766 3.763407e-02 0.087318 NaN
Tnr 26.722797 2.795890e-02 0.091476 NaN
Tmem27 17.364475 1.123328e-141 0.981289 NaN
Slc38a5 15.393972 4.940813e-60 0.920998 NaN
Pcsk1n 12.127839 4.762578e-59 0.997921 NaN
Gast 9.842274 1.419605e-32 0.538462 NaN
Cpe 7.672148 1.079724e-40 1.000000 NaN
Pcsk2 7.646369 6.491126e-63 0.846154 NaN
Gpx3 7.507235 1.191563e-82 0.937630 NaN
Meis2 6.603501 2.217035e-78 0.970894 NaN
Peg10 6.532975 2.869438e-65 0.700624 NaN
Ssr2 6.296807 1.585608e-69 0.985447 NaN
Ppy 6.134921 1.251811e-15 0.378378 NaN
Tmsb15l 5.803368 2.070066e-73 0.711019 NaN
── DEG drivers: Beta vs progenitor ──
Significant: 830 (up: 422, down: 408)
gene logfoldchange pval_adj pct_fate pct_progenitor
Iapp 185.345184 6.106240e-152 0.944162 NaN
Pyy 139.123322 1.052978e-85 0.962775 NaN
Ins1 99.108521 3.512617e-47 0.582064 NaN
Ins2 47.560135 3.515790e-67 0.610829 NaN
Nnat 42.420864 2.029962e-103 0.825719 NaN
Rbp4 41.067871 6.750219e-103 0.996616 NaN
P2ry1 27.845049 4.690784e-06 0.164129 NaN
Fmo1 26.452681 3.894453e-02 0.081218 NaN
Plppr1 26.415466 3.894453e-02 0.081218 NaN
Gnas 26.366089 3.362044e-60 1.000000 NaN
Ttr 22.955372 6.047775e-52 0.994924 NaN
Dlk1 12.507628 4.599450e-97 0.966159 NaN
Sec61b 12.047029 6.698179e-114 1.000000 NaN
Npy 12.042974 1.086211e-07 0.191201 NaN
Ppp1r1a 11.322408 1.013795e-94 0.712352 NaN
Pcsk2 9.181964 2.013853e-127 0.984772 NaN
Calr 8.696516 3.976336e-95 0.959391 NaN
Rpl10 8.650661 6.788600e-35 1.000000 NaN
Hspa5 8.530251 2.027799e-109 0.969543 NaN
Sdf2l1 7.616785 3.683485e-85 0.747885 NaN
── DEG drivers: Delta vs progenitor ──
Significant: 386 (up: 156, down: 230)
gene logfoldchange pval_adj pct_fate pct_progenitor
Pyy 484.160583 5.419924e-34 1.000000 NaN
Sst 332.010773 9.905999e-30 0.871429 NaN
Rbp4 194.580841 2.024127e-37 1.000000 NaN
Iapp 102.586014 2.319858e-35 0.985714 NaN
Ptprz1 28.902637 3.369656e-03 0.257143 NaN
Cd24a 20.200207 1.633998e-31 1.000000 NaN
Ppy 18.444256 6.494096e-07 0.485714 NaN
Hhex 16.970627 6.566081e-31 0.985714 NaN
Dlk1 16.967428 4.469675e-28 1.000000 NaN
Rpl10 13.510839 4.188939e-13 1.000000 NaN
Ssr2 10.061992 2.442933e-24 1.000000 NaN
Gpx3 9.890513 3.689698e-25 0.985714 NaN
Isl1 9.400220 3.580594e-12 1.000000 NaN
Hadh 9.289947 7.724100e-31 0.971429 NaN
Arg1 8.830122 9.905999e-30 0.900000 NaN
Pcsk2 8.306222 1.427407e-20 0.928571 NaN
Ttr 7.740336 4.406957e-03 0.971429 NaN
Spock3 7.235566 4.338918e-05 0.342857 NaN
Mest 6.525658 6.399791e-18 0.714286 NaN
Igfbp7 6.383225 4.517170e-17 0.685714 NaN
── DEG drivers: Epsilon vs progenitor ──
Significant: 518 (up: 266, down: 252)
gene logfoldchange pval_adj pct_fate pct_progenitor
Ghrl 495.486511 3.376669e-72 1.000000 NaN
Pyy 81.501137 3.980708e-18 0.873239 NaN
Rbp4 74.675728 6.843394e-44 0.992958 NaN
Lrpprc 22.582724 8.565872e-16 0.873239 NaN
Cdkn1a 21.258612 4.533111e-15 0.887324 NaN
Gcg 20.178659 1.772858e-04 0.338028 NaN
Iapp 18.956543 1.029570e-23 0.760563 NaN
Isl1 18.905531 3.368229e-35 0.992958 NaN
Ttr 16.562450 4.734949e-04 0.929577 NaN
Maged2 15.220592 1.711104e-41 0.943662 NaN
Hspa5 9.455750 8.303204e-39 0.957746 NaN
Tmem27 8.894045 2.939794e-24 0.739437 NaN
Calr 8.733923 2.532672e-39 0.950704 NaN
Ssr2 7.860883 2.188520e-39 0.992958 NaN
Arg1 7.192320 3.382770e-34 0.732394 NaN
Rpl10 7.168509 2.855208e-09 1.000000 NaN
Slc38a5 7.034479 3.542731e-11 0.838028 NaN
Ccnd2 6.007015 4.025146e-26 0.661972 NaN
Acsl1 5.986801 1.903084e-05 0.267606 NaN
Anpep 5.644922 5.435417e-36 0.802817 NaN
Top DEG drivers (top 5 per fate):
[29]:
| fate | gene | logfoldchange | pval_adj | pct_fate | pct_progenitor | |
|---|---|---|---|---|---|---|
| 0 | Alpha | Pyy | 209.768677 | 1.181761e-93 | 0.968815 | NaN |
| 1 | Alpha | Gcg | 209.614731 | 9.196179e-64 | 0.656965 | NaN |
| 2 | Alpha | Iapp | 99.370232 | 4.065913e-99 | 0.854470 | NaN |
| 3 | Alpha | Ttr | 67.718864 | 2.617159e-103 | 1.000000 | NaN |
| 4 | Alpha | Gnas | 34.805965 | 2.937512e-52 | 1.000000 | NaN |
| 5 | Beta | Iapp | 185.345184 | 6.106240e-152 | 0.944162 | NaN |
| 6 | Beta | Pyy | 139.123322 | 1.052978e-85 | 0.962775 | NaN |
| 7 | Beta | Ins1 | 99.108521 | 3.512617e-47 | 0.582064 | NaN |
| 8 | Beta | Ins2 | 47.560135 | 3.515790e-67 | 0.610829 | NaN |
| 9 | Beta | Nnat | 42.420864 | 2.029962e-103 | 0.825719 | NaN |
| 10 | Delta | Pyy | 484.160583 | 5.419924e-34 | 1.000000 | NaN |
| 11 | Delta | Sst | 332.010773 | 9.905999e-30 | 0.871429 | NaN |
| 12 | Delta | Rbp4 | 194.580841 | 2.024127e-37 | 1.000000 | NaN |
| 13 | Delta | Iapp | 102.586014 | 2.319858e-35 | 0.985714 | NaN |
| 14 | Delta | Ptprz1 | 28.902637 | 3.369656e-03 | 0.257143 | NaN |
| 15 | Epsilon | Ghrl | 495.486511 | 3.376669e-72 | 1.000000 | NaN |
| 16 | Epsilon | Pyy | 81.501137 | 3.980708e-18 | 0.873239 | NaN |
| 17 | Epsilon | Rbp4 | 74.675728 | 6.843394e-44 | 0.992958 | NaN |
| 18 | Epsilon | Tnr | 26.569187 | 2.522357e-01 | 0.091549 | NaN |
| 19 | Epsilon | Hpgds | 26.106819 | 4.514074e-01 | 0.070423 | NaN |
11.3 Velocity-fate correlation drivers¶
get_velocity_fate_drivers() computes the Spearman correlation between each gene’s velocity and the per-cell fate affinity score for each fate arm. Genes with high positive correlation are being upregulated specifically as cells commit to that fate — a stronger signal than mean velocity alone, because it filters out genes that are fast everywhere.
Requires score(cell_level=True) to have been called first (needs result.cell_scores).
[30]:
# Velocity-fate correlation drivers
# Correlates each gene's velocity with per-cell fate affinity scores
# (requires cell_level=True in score())
try:
vf_drivers = scorer.get_velocity_fate_drivers(
result,
n_top_genes=20,
pval_threshold=0.05,
)
print("Top velocity-fate drivers (top 5 per fate):")
display(_stack_drivers(
vf_drivers, n_per_fate=5,
cols=["gene", "spearman_r", "pval_adj", "mean_velocity", "delta_velocity"],
))
except ValueError as e:
print(f"Skipped: {e}")
print("Tip: call scorer.score(cell_level=True) first.")
── Velocity-fate drivers: Alpha (top 20, sorted by Spearman r) ──
Significant (FDR < 0.05): 683 / 2000
rank gene spearman_r pval_adj mean_velocity
1 Adam23 0.332275 2.726568e-46 0.035280
2 Klb 0.309560 6.025260e-40 0.045717
3 Tmem206 0.265787 4.237692e-29 0.099139
4 Notch2 0.257026 2.741812e-27 0.073804
5 Sorcs2 0.235934 5.877863e-23 0.037468
6 Rasgrf2 0.235511 6.661376e-23 0.025164
7 Epha4 0.235026 7.813431e-23 0.052760
8 Megf11 0.228079 1.824037e-21 0.007886
9 Sgce 0.224234 9.184396e-21 0.052389
10 Kif2c 0.223713 1.097454e-20 0.083291
11 Sel1l 0.223152 1.274883e-20 0.055911
12 Bst2 0.222681 1.434448e-20 0.190410
13 Ctsb 0.219757 4.981190e-20 -0.068126
14 Gabrb3 0.217725 1.071893e-19 0.055429
15 Tmem51 0.214352 4.378911e-19 0.109053
16 Fam234a 0.214279 4.378911e-19 0.044228
17 1700011H14Rik 0.212987 7.086263e-19 0.285092
18 Gch1 0.211215 1.369510e-18 0.472799
19 Cdh11 0.211206 1.369510e-18 0.021027
20 Zcchc16 0.210129 2.081817e-18 0.022147
── Velocity-fate drivers: Beta (top 20, sorted by Spearman r) ──
Significant (FDR < 0.05): 924 / 2000
rank gene spearman_r pval_adj mean_velocity
1 Gng12 0.687204 4.410440e-259 0.364720
2 Nnat 0.675808 1.430221e-247 4.051527
3 Sdk2 0.571355 3.286960e-160 0.011898
4 Dact2 0.559535 2.499373e-152 0.045325
5 Ap1s2 0.549641 5.731258e-146 0.087439
6 Vat1l 0.548981 1.264673e-145 0.064852
7 Sphkap 0.541680 4.498258e-141 0.119721
8 Mxd4 0.515216 2.594995e-125 -0.069785
9 Slc2a2 0.511380 2.850628e-123 0.166863
10 Nol4 0.506439 1.524017e-120 0.096487
11 Shroom3 0.501075 1.259699e-117 -0.008355
12 Zbtb7c 0.497687 7.505370e-116 0.024958
13 Ins2 0.488525 5.173891e-111 3.689225
14 Igfbpl1 0.474913 3.992365e-104 0.268164
15 Slc7a14 0.461875 7.903311e-98 0.072172
16 Ptprz1 0.459658 8.304841e-97 0.021035
17 Ccnd2 0.458424 3.072975e-96 0.365967
18 Gc 0.455471 7.272239e-95 0.137919
19 Ghr 0.454304 2.464647e-94 0.248989
20 Slc30a8 0.449188 5.200257e-92 0.450795
── Velocity-fate drivers: Delta (top 20, sorted by Spearman r) ──
Significant (FDR < 0.05): 685 / 2000
rank gene spearman_r pval_adj mean_velocity
1 Zbtb20 0.303609 1.783641e-38 0.166360
2 Fndc3a 0.293026 9.165943e-36 -0.005701
3 Pbld2 0.258827 1.428849e-27 0.038566
4 Vat1l 0.252901 2.293744e-26 0.065950
5 Trim35 0.250792 5.950762e-26 0.184455
6 Hap1 0.248696 1.537300e-25 0.031227
7 Arx 0.245404 7.175120e-25 -0.018569
8 Asb4 0.236969 3.565400e-23 -0.003783
9 Vps37a 0.227608 2.126823e-21 -0.083059
10 Chd3 0.223959 9.343843e-21 -0.079594
11 Snap25 0.222951 1.330732e-20 0.048876
12 Mia3 0.221878 2.046525e-20 0.047561
13 Ufm1 0.217722 1.073379e-19 -0.142988
14 Stard10 0.214950 3.289204e-19 -0.088858
15 BC048546 0.214442 3.949377e-19 -0.140049
16 Pon2 0.208136 4.756439e-18 -0.013737
17 Tmx1 0.203896 2.536844e-17 -0.220162
18 Cpm 0.202291 4.706904e-17 0.038128
19 Gcsh 0.201279 6.868679e-17 -0.086339
20 Hagh 0.198569 1.954275e-16 0.038047
── Velocity-fate drivers: Epsilon (top 20, sorted by Spearman r) ──
Significant (FDR < 0.05): 924 / 2000
rank gene spearman_r pval_adj mean_velocity
1 Rab27a 0.514252 8.184805e-125 0.055813
2 Pdk2 0.512925 4.186366e-124 0.051594
3 Sorcs1 0.499450 8.951898e-117 0.005206
4 Il17re 0.485971 1.042002e-109 -0.002558
5 Auts2 0.484087 9.271443e-109 0.256760
6 Pycr2 0.474232 8.299458e-104 -0.452137
7 Nusap1 0.467321 2.024908e-100 0.014434
8 Mnx1 0.465603 1.321285e-99 -0.004751
9 Chga 0.460968 2.055227e-97 -2.422998
10 Npepl1 0.449508 3.960569e-92 -0.043343
11 Gm609 0.449416 4.222258e-92 0.024659
12 Efcab10 0.448844 7.240713e-92 -0.045064
13 Gsto1 0.445875 1.588429e-90 -0.086271
14 Igf1r 0.445424 2.468092e-90 -0.019749
15 Eps8l2 0.442219 6.679705e-89 0.126253
16 Kcnb2 0.441447 1.439386e-88 0.029580
17 Cbfa2t2 0.432747 9.568963e-85 0.159800
18 Fam213a 0.432126 1.687532e-84 0.043521
19 Josd2 0.428882 4.017044e-83 0.046612
20 Kif20a 0.428003 9.327305e-83 0.081612
Top velocity-fate drivers (top 5 per fate):
| fate | gene | spearman_r | pval_adj | mean_velocity | delta_velocity | |
|---|---|---|---|---|---|---|
| 0 | Alpha | Adam23 | 0.332275 | 2.726568e-46 | 0.035280 | 0.026601 |
| 1 | Alpha | Klb | 0.309560 | 6.025260e-40 | 0.045717 | 0.037019 |
| 2 | Alpha | Tmem206 | 0.265787 | 4.237692e-29 | 0.099139 | 0.149552 |
| 3 | Alpha | Notch2 | 0.257026 | 2.741812e-27 | 0.073804 | 0.069658 |
| 4 | Alpha | Sorcs2 | 0.235934 | 5.877863e-23 | 0.037468 | 0.026698 |
| 5 | Beta | Gng12 | 0.687204 | 4.410440e-259 | 0.364720 | 0.065286 |
| 6 | Beta | Nnat | 0.675808 | 1.430221e-247 | 4.051527 | 3.631306 |
| 7 | Beta | Sdk2 | 0.571355 | 3.286960e-160 | 0.011898 | 0.011638 |
| 8 | Beta | Dact2 | 0.559535 | 2.499373e-152 | 0.045325 | 0.018165 |
| 9 | Beta | Ap1s2 | 0.549641 | 5.731258e-146 | 0.087439 | 0.024644 |
| 10 | Delta | Zbtb20 | 0.303609 | 1.783641e-38 | 0.166360 | -0.036171 |
| 11 | Delta | Fndc3a | 0.293026 | 9.165943e-36 | -0.005701 | -0.049914 |
| 12 | Delta | Pbld2 | 0.258827 | 1.428849e-27 | 0.038566 | 0.025424 |
| 13 | Delta | Vat1l | 0.252901 | 2.293744e-26 | 0.065950 | 0.013837 |
| 14 | Delta | Trim35 | 0.250792 | 5.950762e-26 | 0.184455 | 0.063726 |
| 15 | Epsilon | Rab27a | 0.514252 | 8.184805e-125 | 0.055813 | 0.007661 |
| 16 | Epsilon | Pdk2 | 0.512925 | 4.186366e-124 | 0.051594 | 0.072639 |
| 17 | Epsilon | Sorcs1 | 0.499450 | 8.951898e-117 | 0.005206 | 0.004127 |
| 18 | Epsilon | Il17re | 0.485971 | 1.042002e-109 | -0.002558 | -0.027604 |
| 19 | Epsilon | Auts2 | 0.484087 | 9.271443e-109 | 0.256760 | 0.302319 |
12. Pathway enrichment¶
get_enrichment() runs over-representation analysis (ORA) on the DEG driver genes for each fate using gseapy (KEGG, GO Biological Process, Reactome).
Requires an internet connection and a valid organism code.
[31]:
# Pathway enrichment per fate
# Note: requires gseapy and internet access for Enrichr API
# Results will be empty with synthetic data (too few significant DEGs)
try:
enrichment = scorer.get_enrichment(
deg_drivers,
organism="mouse", # "mouse" or "human"
pval_threshold=0.05,
logfc_threshold=0.25,
plot=True, # dot plots saved per fate
n_top_pathways=15,
)
# Single compact summary: top 3 pathways per (fate, direction)
enrich_df = _stack_enrichment(enrichment, n_per_group=3)
if not enrich_df.empty:
display(enrich_df[["fate", "direction", "Gene_set", "Term", "Overlap", "Adjusted P-value"]])
else:
print("(No enriched terms — typical with small / synthetic datasets.)")
except ImportError:
print("gseapy not installed. Run: pip install gseapy")
except Exception as e:
print(f"Enrichment skipped: {e}")
print("(Expected with synthetic data — use real data for meaningful results.)")
============================================================
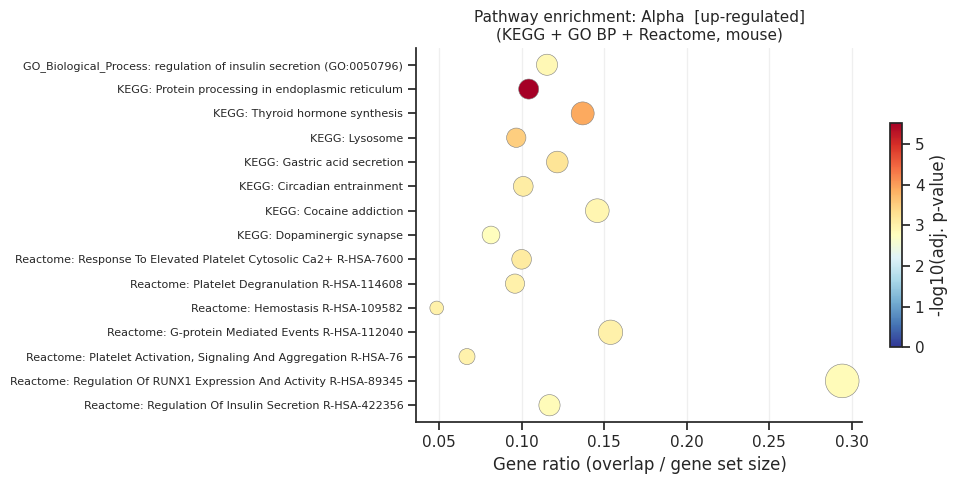
Pathway enrichment: Alpha
Gene sets: ['KEGG_2019_Mouse', 'GO_Biological_Process_2021', 'Reactome_2022']
Up-regulated genes : 376
Down-regulated genes: 385
============================================================
[up] Significant terms: 121
Gene_set Term Overlap Adjusted P-value
KEGG_2019_Mouse Protein processing in endoplasmic reticulum 17/163 0.000003
KEGG_2019_Mouse Thyroid hormone synthesis 10/73 0.000127
KEGG_2019_Mouse Lysosome 12/124 0.000313
KEGG_2019_Mouse Gastric acid secretion 9/74 0.000600
Reactome_2022 Response To Elevated Platelet Cytosolic Ca2+ R-HSA-76005 13/130 0.000783
KEGG_2019_Mouse Circadian entrainment 10/99 0.000825
Reactome_2022 Platelet Degranulation R-HSA-114608 12/125 0.000966
Reactome_2022 Hemostasis R-HSA-109582 28/576 0.000966
Reactome_2022 G-protein Mediated Events R-HSA-112040 8/52 0.000966
Reactome_2022 Platelet Activation, Signaling And Aggregation R-HSA-76002 17/254 0.001008
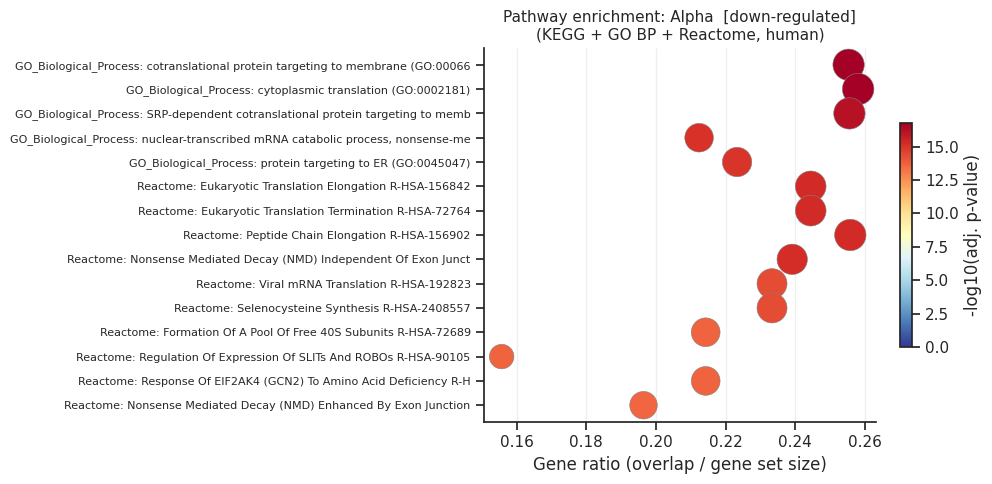
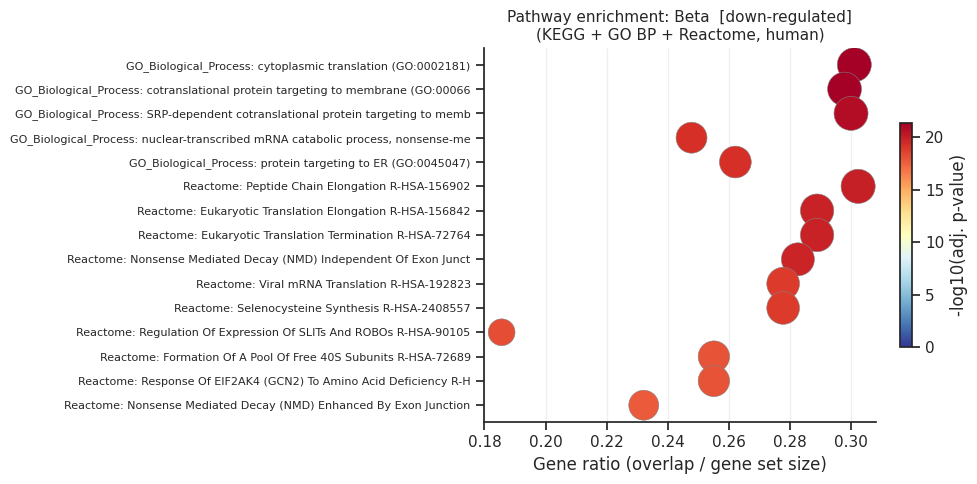
[down] Significant terms: 252
Gene_set Term Overlap Adjusted P-value
GO_Biological_Process_2021 cotranslational protein targeting to membrane (GO:0006613) 24/94 1.627570e-17
GO_Biological_Process_2021 cytoplasmic translation (GO:0002181) 24/93 1.627570e-17
GO_Biological_Process_2021 SRP-dependent cotranslational protein targeting to membrane (GO:0006614) 23/90 6.790532e-17
Reactome_2022 Eukaryotic Translation Elongation R-HSA-156842 22/90 5.465181e-16
Reactome_2022 Eukaryotic Translation Termination R-HSA-72764 22/90 5.465181e-16
Reactome_2022 Peptide Chain Elongation R-HSA-156902 22/86 5.465181e-16
Reactome_2022 Nonsense Mediated Decay (NMD) Independent Of Exon Junction Complex (EJC) R-HSA-975956 22/92 6.859847e-16
GO_Biological_Process_2021 nuclear-transcribed mRNA catabolic process, nonsense-mediated decay (GO:0000184) 24/113 8.686504e-16
GO_Biological_Process_2021 protein targeting to ER (GO:0045047) 23/103 1.101897e-15
Reactome_2022 Viral mRNA Translation R-HSA-192823 21/90 4.696474e-15
============================================================
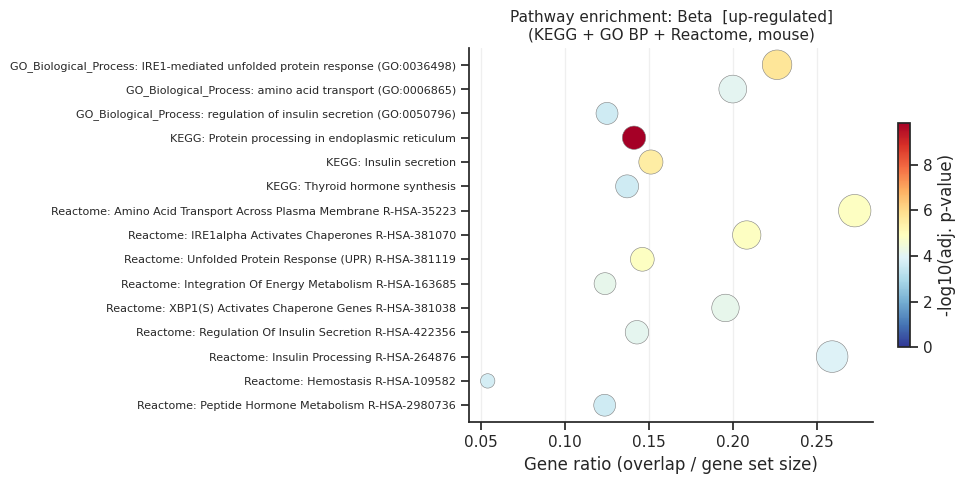
Pathway enrichment: Beta
Gene sets: ['KEGG_2019_Mouse', 'GO_Biological_Process_2021', 'Reactome_2022']
Up-regulated genes : 422
Down-regulated genes: 408
============================================================
[up] Significant terms: 121
Gene_set Term Overlap Adjusted P-value
KEGG_2019_Mouse Protein processing in endoplasmic reticulum 23/163 1.407868e-10
GO_Biological_Process_2021 IRE1-mediated unfolded protein response (GO:0036498) 12/53 1.863342e-06
KEGG_2019_Mouse Insulin secretion 13/86 3.415612e-06
Reactome_2022 Amino Acid Transport Across Plasma Membrane R-HSA-352230 9/33 1.321921e-05
Reactome_2022 IRE1alpha Activates Chaperones R-HSA-381070 10/48 1.321921e-05
Reactome_2022 Unfolded Protein Response (UPR) R-HSA-381119 13/89 1.321921e-05
Reactome_2022 Integration Of Energy Metabolism R-HSA-163685 13/105 6.483513e-05
Reactome_2022 XBP1(S) Activates Chaperone Genes R-HSA-381038 9/46 6.628341e-05
Reactome_2022 Regulation Of Insulin Secretion R-HSA-422356 11/77 8.152971e-05
GO_Biological_Process_2021 amino acid transport (GO:0006865) 10/50 8.704602e-05
[down] Significant terms: 245
Gene_set Term Overlap Adjusted P-value
GO_Biological_Process_2021 cytoplasmic translation (GO:0002181) 28/93 4.333388e-22
GO_Biological_Process_2021 cotranslational protein targeting to membrane (GO:0006613) 28/94 4.333388e-22
GO_Biological_Process_2021 SRP-dependent cotranslational protein targeting to membrane (GO:0006614) 27/90 1.679608e-21
Reactome_2022 Peptide Chain Elongation R-HSA-156902 26/86 1.148227e-20
Reactome_2022 Eukaryotic Translation Elongation R-HSA-156842 26/90 1.428872e-20
Reactome_2022 Eukaryotic Translation Termination R-HSA-72764 26/90 1.428872e-20
Reactome_2022 Nonsense Mediated Decay (NMD) Independent Of Exon Junction Complex (EJC) R-HSA-975956 26/92 2.014986e-20
GO_Biological_Process_2021 nuclear-transcribed mRNA catabolic process, nonsense-mediated decay (GO:0000184) 28/113 5.547475e-20
GO_Biological_Process_2021 protein targeting to ER (GO:0045047) 27/103 5.547475e-20
Reactome_2022 Viral mRNA Translation R-HSA-192823 25/90 1.461345e-19
============================================================
Pathway enrichment: Delta
Gene sets: ['KEGG_2019_Mouse', 'GO_Biological_Process_2021', 'Reactome_2022']
Up-regulated genes : 156
Down-regulated genes: 230
============================================================
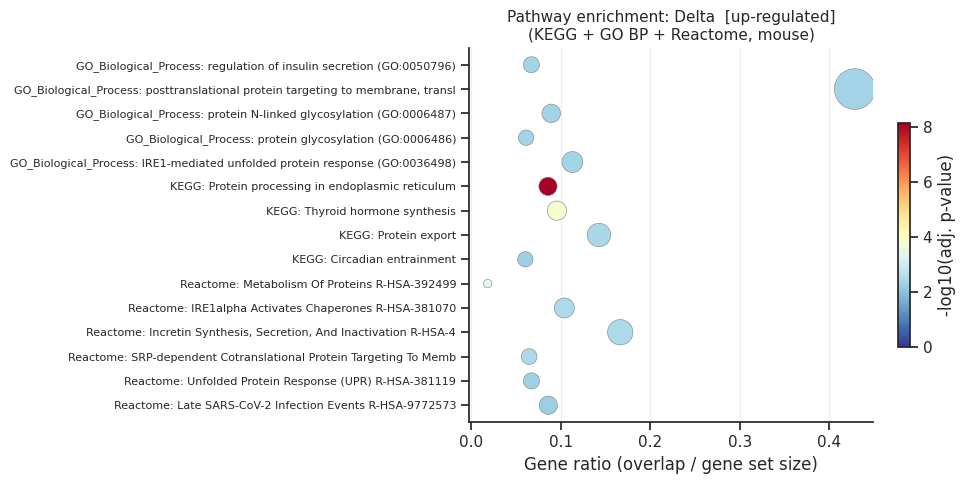
[up] Significant terms: 41
Gene_set Term Overlap Adjusted P-value
KEGG_2019_Mouse Protein processing in endoplasmic reticulum 14/163 6.690234e-09
KEGG_2019_Mouse Thyroid hormone synthesis 7/73 1.429359e-04
Reactome_2022 Metabolism Of Proteins R-HSA-392499 35/1890 3.776032e-04
Reactome_2022 IRE1alpha Activates Chaperones R-HSA-381070 5/48 3.289099e-03
Reactome_2022 Incretin Synthesis, Secretion, And Inactivation R-HSA-400508 4/24 3.289099e-03
Reactome_2022 SRP-dependent Cotranslational Protein Targeting To Membrane R-HSA-1799339 7/108 3.289099e-03
KEGG_2019_Mouse Protein export 4/28 3.697039e-03
GO_Biological_Process_2021 regulation of insulin secretion (GO:0050796) 7/104 4.368748e-03
GO_Biological_Process_2021 posttranslational protein targeting to membrane, translocation (GO:0031204) 3/7 4.368748e-03
GO_Biological_Process_2021 protein N-linked glycosylation (GO:0006487) 6/67 4.368748e-03
============================================================
Pathway enrichment: Epsilon
Gene sets: ['KEGG_2019_Mouse', 'GO_Biological_Process_2021', 'Reactome_2022']
Up-regulated genes : 266
Down-regulated genes: 252
============================================================
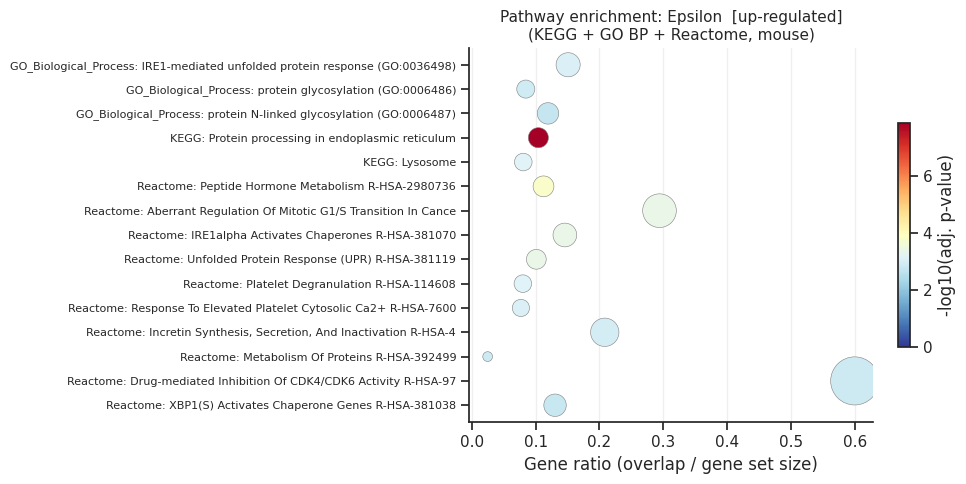
[up] Significant terms: 47
Gene_set Term Overlap Adjusted P-value
KEGG_2019_Mouse Protein processing in endoplasmic reticulum 17/163 1.392463e-08
Reactome_2022 Peptide Hormone Metabolism R-HSA-2980736 10/89 1.607304e-04
Reactome_2022 Aberrant Regulation Of Mitotic G1/S Transition In Cancer Due To RB1 Defects R-HSA-9659787 5/17 4.275598e-04
Reactome_2022 IRE1alpha Activates Chaperones R-HSA-381070 7/48 4.275598e-04
Reactome_2022 Unfolded Protein Response (UPR) R-HSA-381119 9/89 4.275598e-04
KEGG_2019_Mouse Lysosome 10/124 6.781878e-04
Reactome_2022 Platelet Degranulation R-HSA-114608 10/125 7.374527e-04
GO_Biological_Process_2021 IRE1-mediated unfolded protein response (GO:0036498) 8/53 8.563951e-04
Reactome_2022 Response To Elevated Platelet Cytosolic Ca2+ R-HSA-76005 10/130 8.709228e-04
Reactome_2022 Incretin Synthesis, Secretion, And Inactivation R-HSA-400508 5/24 1.076303e-03
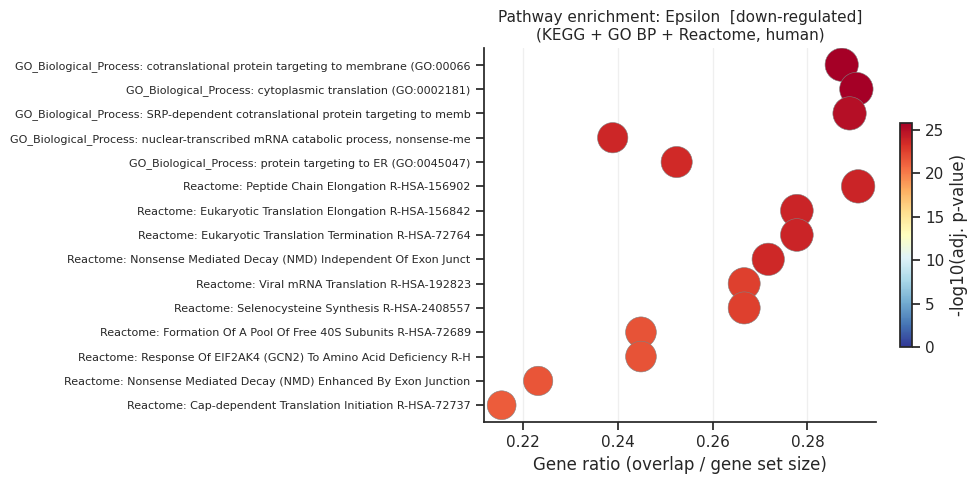
[down] Significant terms: 361
Gene_set Term Overlap Adjusted P-value
GO_Biological_Process_2021 cotranslational protein targeting to membrane (GO:0006613) 27/94 1.419635e-26
GO_Biological_Process_2021 cytoplasmic translation (GO:0002181) 27/93 1.419635e-26
GO_Biological_Process_2021 SRP-dependent cotranslational protein targeting to membrane (GO:0006614) 26/90 9.147683e-26
Reactome_2022 Peptide Chain Elongation R-HSA-156902 25/86 1.231498e-24
Reactome_2022 Eukaryotic Translation Elongation R-HSA-156842 25/90 1.482618e-24
Reactome_2022 Eukaryotic Translation Termination R-HSA-72764 25/90 1.482618e-24
GO_Biological_Process_2021 nuclear-transcribed mRNA catabolic process, nonsense-mediated decay (GO:0000184) 27/113 1.781313e-24
Reactome_2022 Nonsense Mediated Decay (NMD) Independent Of Exon Junction Complex (EJC) R-HSA-975956 25/92 2.059459e-24
GO_Biological_Process_2021 protein targeting to ER (GO:0045047) 26/103 2.772652e-24
Reactome_2022 Viral mRNA Translation R-HSA-192823 24/90 2.428940e-23
| fate | direction | Gene_set | Term | Overlap | Adjusted P-value | |
|---|---|---|---|---|---|---|
| 0 | Alpha | up | KEGG_2019_Mouse | Protein processing in endoplasmic reticulum | 17/163 | 3.014548e-06 |
| 1 | Alpha | up | KEGG_2019_Mouse | Thyroid hormone synthesis | 10/73 | 1.267707e-04 |
| 2 | Alpha | up | KEGG_2019_Mouse | Lysosome | 12/124 | 3.133244e-04 |
| 3 | Alpha | down | GO_Biological_Process_2021 | cotranslational protein targeting to membrane ... | 24/94 | 1.627570e-17 |
| 4 | Alpha | down | GO_Biological_Process_2021 | cytoplasmic translation (GO:0002181) | 24/93 | 1.627570e-17 |
| ... | ... | ... | ... | ... | ... | ... |
| 16 | Epsilon | up | Reactome_2022 | Peptide Hormone Metabolism R-HSA-2980736 | 10/89 | 1.607304e-04 |
| 17 | Epsilon | up | Reactome_2022 | Aberrant Regulation Of Mitotic G1/S Transition... | 5/17 | 4.275598e-04 |
| 18 | Epsilon | down | GO_Biological_Process_2021 | cotranslational protein targeting to membrane ... | 27/94 | 1.419635e-26 |
| 19 | Epsilon | down | GO_Biological_Process_2021 | cytoplasmic translation (GO:0002181) | 27/93 | 1.419635e-26 |
| 20 | Epsilon | down | GO_Biological_Process_2021 | SRP-dependent cotranslational protein targetin... | 26/90 | 9.147683e-26 |
21 rows × 6 columns
[32]:
# ── Custom gene sets ──────────────────────────────────────────────────────
# You can specify any Enrichr library:
# https://maayanlab.cloud/Enrichr/#libraries
custom_gene_sets = [
'KEGG_2019_Mouse',
'GO_Biological_Process_2021',
'GO_Molecular_Function_2021',
'Reactome_2022',
'WikiPathway_2021_Mouse',
]
# enrichment = scorer.get_enrichment(
# deg_drivers,
# gene_sets=custom_gene_sets,
# organism='mouse',
# )
print("Custom gene sets configured (commented out to avoid API call with synthetic data).")
print(f"Available sets: {custom_gene_sets}")
Custom gene sets configured (commented out to avoid API call with synthetic data).
Available sets: ['KEGG_2019_Mouse', 'GO_Biological_Process_2021', 'GO_Molecular_Function_2021', 'Reactome_2022', 'WikiPathway_2021_Mouse']
[33]:
# Export enrichment tables to CSV
try:
scCS.export_enrichment_tables(enrichment, output_dir="enrichment_results/")
print("Tables saved to enrichment_results/")
except Exception as e:
print(f"Export skipped: {e}")
Saved: enrichment_results/enrichment_Alpha_up.csv
Saved: enrichment_results/enrichment_Alpha_down.csv
Saved: enrichment_results/enrichment_Beta_up.csv
Saved: enrichment_results/enrichment_Beta_down.csv
Saved: enrichment_results/enrichment_Delta_up.csv
Saved: enrichment_results/enrichment_Epsilon_up.csv
Saved: enrichment_results/enrichment_Epsilon_down.csv
Tables saved to enrichment_results/
13. Expression trends along fate arms¶
plot_expression_trends() shows how gene expression changes along each fate arm, ordered by a differentiation metric.
The x_axis parameter controls the ordering:
'affinity'(default) — per-cell fate affinity score'pseudotime'— subset-local pseudotime'radial_distance'— distance from origin in the star embedding
[34]:
# Pick a few marker genes
genes_of_interest = ["Ins1", "Gcg", "Sst", "Ppy"] # Beta, Alpha, Delta, Epsilon markers
# Expression trends ordered by fate affinity
# Signature: plot_expression_trends(adata_sub, result, genes, x_axis=...)
try:
fig = scCS.plot_expression_trends(
scorer.adata_sub,
result,
genes=genes_of_interest,
x_axis="affinity", # or 'pseudotime' or 'radial_distance'
figsize=(12, 8),
)
plt.tight_layout()
plt.show()
except Exception as e:
print(f"Expression trends skipped: {e}")
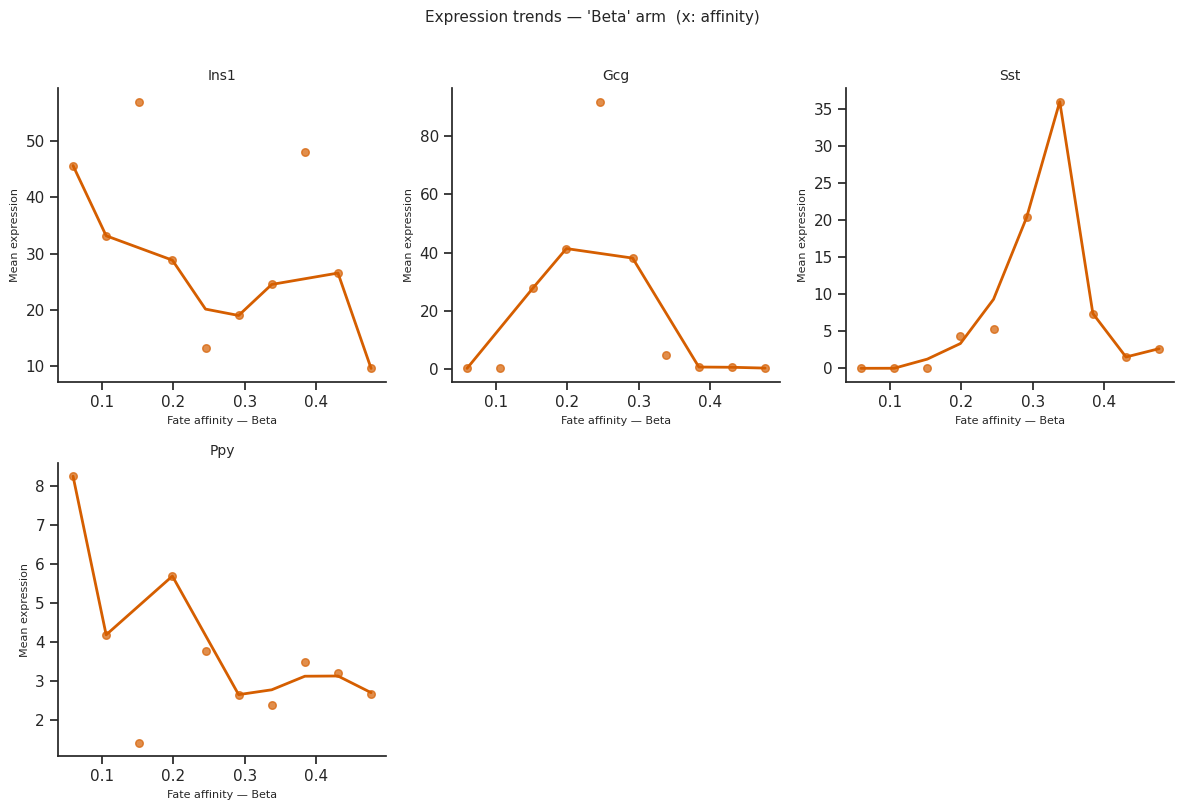
[35]:
# Ordered by pseudotime
try:
fig = scCS.plot_expression_trends(
scorer.adata_sub,
result,
genes=genes_of_interest,
x_axis="pseudotime",
figsize=(12, 8),
)
plt.tight_layout()
plt.show()
except Exception as e:
print(f"Expression trends skipped: {e}")
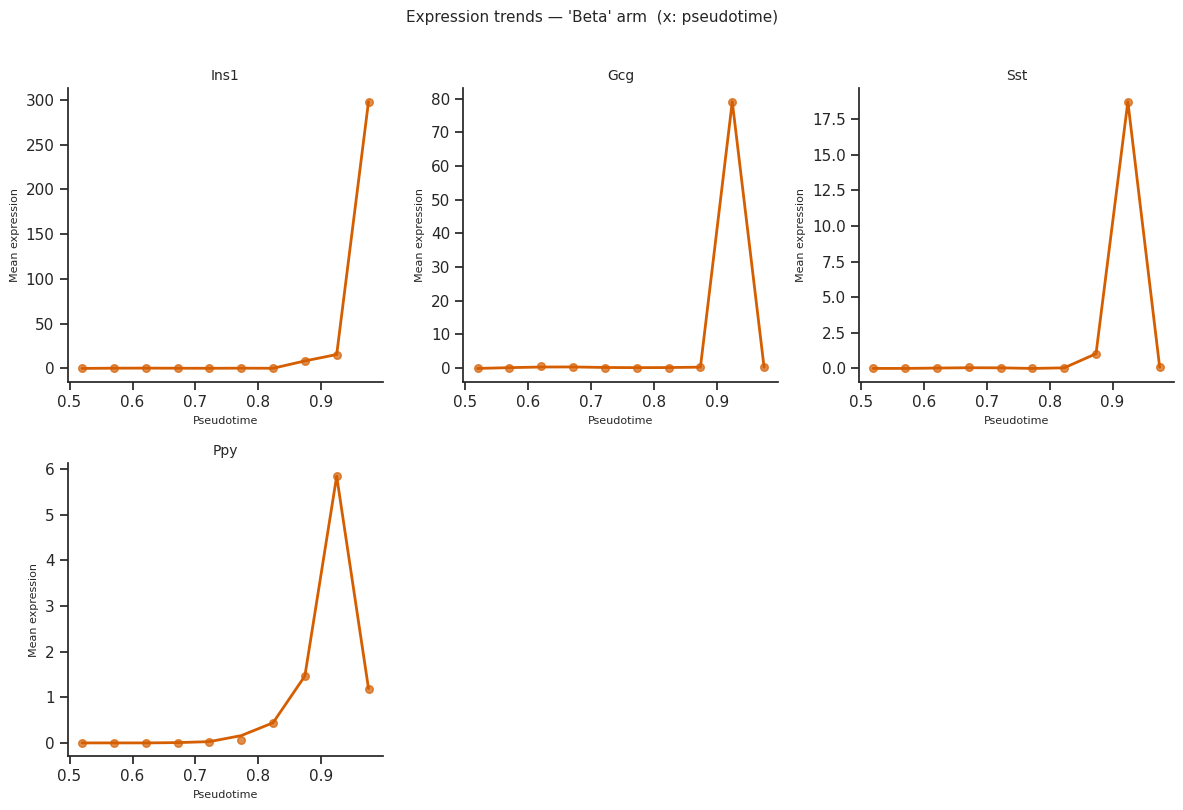
[36]:
obs_key = "clusters" # <-- change to your obs column
color_map = dict(zip(
adata.obs[obs_key].cat.categories,
adata.uns[f"{obs_key}_colors"],
))
print(color_map)
{'Ductal': '#8fbc8f', 'Ngn3 low EP': '#f4a460', 'Ngn3 high EP': '#fdbf6f', 'Pre-endocrine': '#ff7f00', 'Beta': '#b2df8a', 'Alpha': '#1f78b4', 'Delta': '#6a3d9a', 'Epsilon': '#cab2d6'}
[37]:
for fate_name in result.fate_names:
fig = scCS.plot_expression_trends(
adata, result,
genes=genes_of_interest,
fate=fate_name,
color_map=color_map,
ncols=4,
)
fig.suptitle(f"Expression trends — {fate_name}", y=1.02)
fig.savefig(f"expression_trends_{fate_name}.svg", bbox_inches="tight")
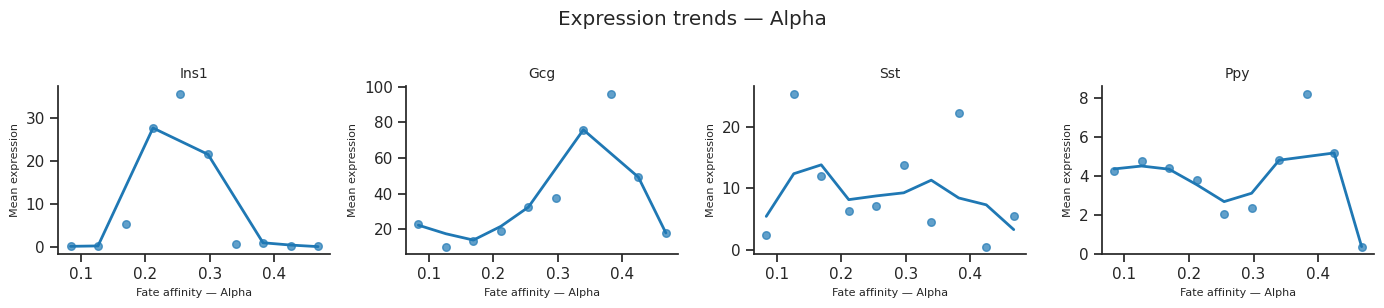
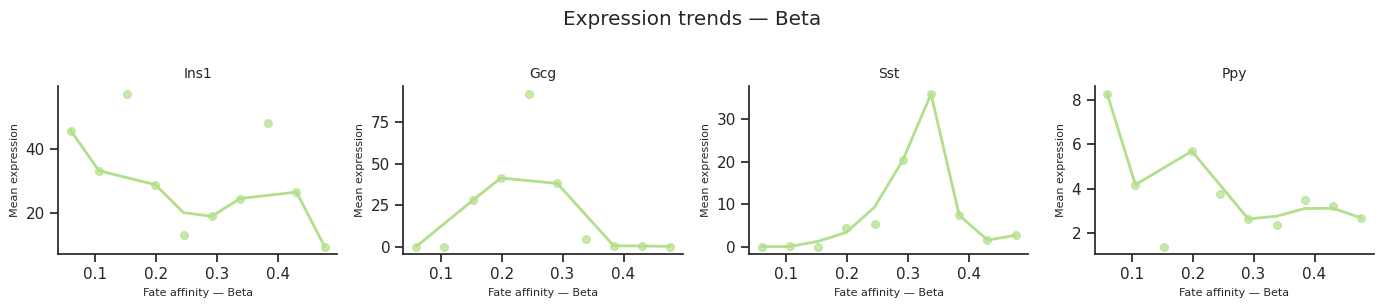
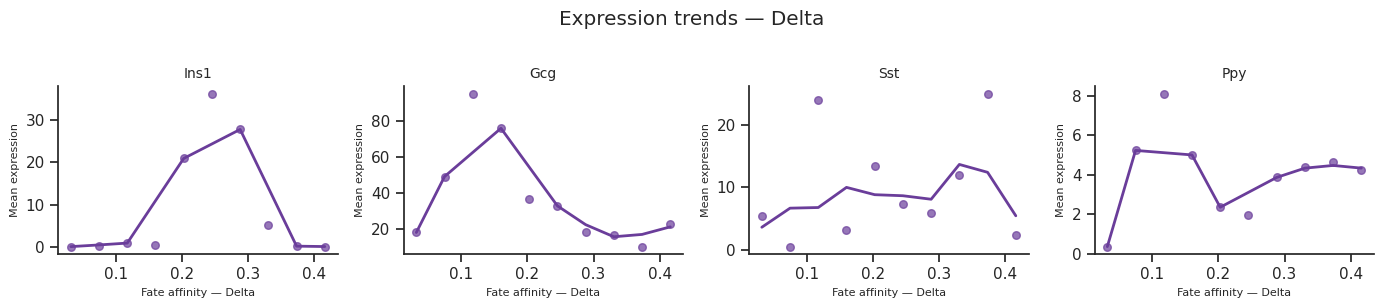
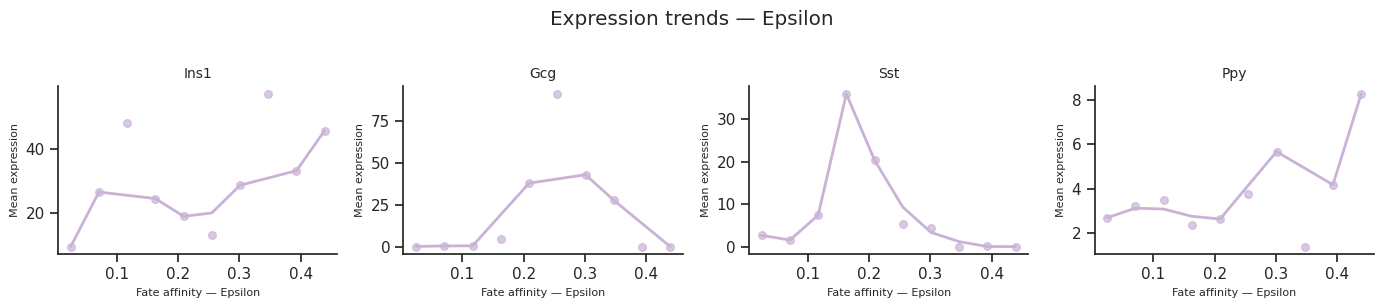
[38]:
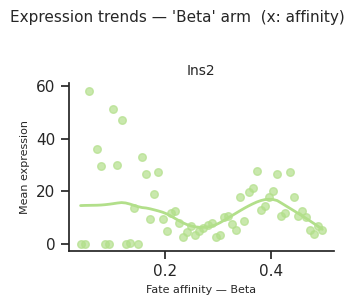
# CellRank-style: cells binned by per-cell CS for a chosen fate,
# mean expression per bin plotted with LOWESS smooth.
# Requires compute_cell_level=True (Cell 2 above).
genes_of_interest = ["Ins2"] # <-- your genes
fig = scCS.plot_expression_trends(
adata,
result,
genes=genes_of_interest,
fate=result.dominant_fate, # or e.g. fate="Activated"
n_bins=60, # bins along CS axis
layer=None, # or None to use adata.X
smooth=True,
smooth_frac=0.4, # LOWESS bandwidth (0–1)
color_map=color_map,
ncols=2,
)
plt.show()
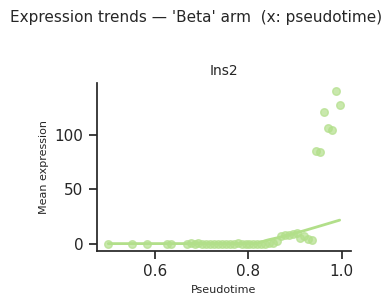
[39]:
fig = scCS.plot_expression_trends(
adata,
result,
genes=genes_of_interest,
fate=result.dominant_fate, # or e.g. fate="Activated"
n_bins=60, # bins along CS axis
layer=None, # or None to use adata.X
smooth=True,
smooth_frac=0.4, # LOWESS bandwidth (0–1)
color_map=color_map,
ncols=2, x_axis="pseudotime",
)
plt.show()
Summary — scCS single-condition workflow¶
import scCS
# 1. Create scorer
scorer = scCS.SingleScorer(
adata,
root="Ductal",
branches=["Alpha", "Beta", "Delta", "Epsilon"],
obs_key="clusters",
)
# 2. Build embedding (fix arm coverage)
scorer.build_embedding(ordering_metric="pseudotime")
scorer.refit_pseudotime(scale_01=True)
# 3. Fit & score
scorer.fit()
result = scorer.score(cell_level=True, k_nn=15, n_bootstrap=500)
print(result.summary())
# 4. Visualize
scorer.plot_star(result)
scorer.plot_rose(result)
scorer.plot_pairwise_cs(result)
scorer.plot_commitment_bar(result)
# 5. Transfer labels to full adata
scorer.transfer_labels(adata, result)
# 6. Driver genes & enrichment
vel_drivers = scorer.get_velocity_drivers()
deg_drivers = scorer.get_deg_drivers()
enrichment = scorer.get_enrichment(deg_drivers, organism="Human")
For multi-condition analysis, see the companion notebooks