scCS Tutorial — Pairwise Condition Comparison¶
Multi-Condition Commitment Score Analysis with PairScorer¶
This notebook demonstrates the multi-condition analysis workflow in scCS v0.7.0.
We artificially split the pancreas dataset into two groups:
“high_velocity” — cells with above-median RNA velocity magnitude (stronger commitment signal)
“low_velocity” — cells with below-median RNA velocity magnitude (weaker commitment signal)
This simulates a treatment effect where one condition drives stronger fate commitment.
Three-tier analysis framework¶
Tier |
What it answers |
Key functions |
|---|---|---|
Tier 1 |
What are the commitment scores per condition? |
|
Tier 2 |
Is the difference statistically significant? |
|
Tier 3 |
Does the trajectory shift between conditions? |
|
Reference¶
Kriukov et al. (2025) Single-cell transcriptome of myeloid cells in response to transplantation of human retinal neurons reveals reversibility of microglial activation
1. Setup and data loading¶
[1]:
%matplotlib inline
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import scanpy as sc
import scvelo as scv
import warnings
warnings.filterwarnings("ignore", category=DeprecationWarning)
warnings.filterwarnings("ignore", category=FutureWarning)
try:
from statsmodels.tools.sm_exceptions import ConvergenceWarning
warnings.filterwarnings("ignore", category=ConvergenceWarning)
except ImportError:
pass
import scCS
print(f"scCS version: {scCS.__version__}")
sc.settings.verbosity = 1
scv.settings.verbosity = 1
# --- Tutorial display helpers (compact tables instead of per-fate loops) ---
pd.set_option("display.max_rows", 25)
pd.set_option("display.max_columns", 15)
pd.set_option("display.width", 160)
def _stack_drivers_by_condition(driver_dicts, n_per_fate=5, cols=None):
"""Stack {condition: {fate: DataFrame}} into one tidy long-form table.
Adds ``condition`` and ``fate`` columns at the front; shows top
``n_per_fate`` rows per (condition, fate) group.
"""
out = []
for cond, fate_dict in driver_dicts.items():
for fate, df in fate_dict.items():
if df is None or len(df) == 0:
continue
block = df.head(n_per_fate).copy()
block.insert(0, "fate", fate)
block.insert(0, "condition", cond)
out.append(block)
if not out:
return pd.DataFrame()
big = pd.concat(out, ignore_index=True)
if cols is not None:
keep = ["condition", "fate"] + [c for c in cols if c in big.columns]
big = big[keep]
return big
def _stack_enrichment_by_condition(enrichment_dicts, n_per_group=3):
"""Flatten {condition: {fate: {direction: df}}} into one tidy table."""
rows = []
for cond, fate_results in enrichment_dicts.items():
for fate, dir_results in fate_results.items():
for direction in ("up", "down"):
df = dir_results.get(direction, None)
if df is None or df.empty:
continue
top = df.head(n_per_group).copy()
top.insert(0, "direction", direction)
top.insert(0, "fate", fate)
top.insert(0, "condition", cond)
rows.append(top)
if not rows:
return pd.DataFrame()
return pd.concat(rows, ignore_index=True)
def _driver_overlap(per_cond_drivers, n_top=10, gene_col="gene"):
"""Build a fate x gene x condition presence matrix from {cond: {fate: df}}."""
rows = []
conds = list(per_cond_drivers.keys())
if not conds:
return pd.DataFrame()
fates = list(next(iter(per_cond_drivers.values())).keys())
for fate in fates:
sets = {}
for cond in conds:
df = per_cond_drivers[cond].get(fate)
if df is None or len(df) == 0 or gene_col not in df.columns:
sets[cond] = set()
else:
sets[cond] = set(df.head(n_top)[gene_col].astype(str))
all_genes = sorted(set.union(*sets.values())) if sets else []
for g in all_genes:
row = {"fate": fate, "gene": g}
for cond in conds:
row[cond] = g in sets[cond]
row["n_conds"] = sum(row[c] for c in conds)
row["shared_all"] = row["n_conds"] == len(conds)
rows.append(row)
return pd.DataFrame(rows)
scCS version: 0.7.4
[2]:
# Load the pancreas dataset
adata = scv.datasets.pancreas()
# Standard preprocessing
import inspect as _inspect
_fn_sig = _inspect.signature(scv.pp.filter_and_normalize).parameters
if "n_top_genes" in _fn_sig:
scv.pp.filter_and_normalize(adata, min_shared_counts=20, n_top_genes=2000)
else:
# scvelo builds without n_top_genes: do filter_and_normalize then HVG.
# Use flavor="cell_ranger" not the default "seurat": seurat passes
# int n_bins to pd.cut on log-dispersions which contain +/-inf for
# low-mean genes — pandas >= 2.2 rejects this with ValueError.
# cell_ranger uses explicit bin edges including +/-inf and is
# robust across pandas/py versions (scCS changelog v0.7.4).
scv.pp.filter_and_normalize(adata, min_shared_counts=20)
sc.pp.highly_variable_genes(
adata, n_top_genes=2000, subset=True, flavor="cell_ranger"
)
sc.pp.neighbors(adata)
scv.pp.moments(adata, n_pcs=None, n_neighbors=None)
print(adata)
AnnData object with n_obs × n_vars = 3696 × 2000
obs: 'clusters_coarse', 'clusters', 'S_score', 'G2M_score', 'initial_size_unspliced', 'initial_size_spliced', 'initial_size', 'n_counts'
var: 'highly_variable_genes', 'gene_count_corr', 'highly_variable', 'means', 'dispersions', 'dispersions_norm'
uns: 'clusters_coarse_colors', 'clusters_colors', 'day_colors', 'neighbors', 'pca', 'hvg'
obsm: 'X_pca', 'X_umap'
layers: 'spliced', 'unspliced', 'Ms', 'Mu'
obsp: 'distances', 'connectivities'
[3]:
# Run RNA velocity (dynamical model)
scv.tl.recover_dynamics(adata, n_jobs=32)
scv.tl.velocity(adata, mode="dynamical")
scv.tl.velocity_graph(adata)
scv.tl.velocity_pseudotime(adata)
print("Velocity computed.")
/home/emil/miniforge3/envs/lab-py312/lib/python3.12/multiprocessing/popen_fork.py:66: DeprecationWarning: This process (pid=4708) is multi-threaded, use of fork() may lead to deadlocks in the child.
self.pid = os.fork()
/home/emil/miniforge3/envs/lab-py312/lib/python3.12/multiprocessing/popen_fork.py:66: DeprecationWarning: This process (pid=4708) is multi-threaded, use of fork() may lead to deadlocks in the child.
self.pid = os.fork()
Velocity computed.
2. Create artificial condition split¶
We split cells into two groups based on their RNA velocity magnitude:
high_velocity — cells with above-median velocity magnitude
low_velocity — cells with below-median velocity magnitude
This simulates a scenario where a treatment (e.g., a drug or genetic perturbation) increases the strength of fate commitment signals.
In a real experiment,
condition_obs_keywould point to a column like'treatment','genotype', or'time_point'already present inadata.obs.
[4]:
# Compute per-cell velocity magnitude from the velocity layer
velocity_matrix = adata.layers["velocity"] # shape: (n_cells, n_genes)
# Use RMS across genes as a per-cell magnitude proxy
vel_magnitude = np.sqrt(np.nanmean(velocity_matrix ** 2, axis=1))
# Split at the median
median_mag = np.median(vel_magnitude)
adata.obs["condition"] = np.where(
vel_magnitude >= median_mag, "high_velocity", "low_velocity"
)
# Also create a fake replicate column (for mixed model demo)
# In practice this would be biological replicates / samples
np.random.seed(42)
adata.obs["sample"] = np.random.choice(
["rep1", "rep2", "rep3"], size=adata.n_obs
)
print("Condition distribution:")
print(adata.obs["condition"].value_counts())
print("\nSample distribution:")
print(adata.obs["sample"].value_counts())
Condition distribution:
condition
high_velocity 1848
low_velocity 1848
Name: count, dtype: int64
Sample distribution:
sample
rep1 1282
rep2 1224
rep3 1190
Name: count, dtype: int64
[5]:
# Visualize the split on UMAP
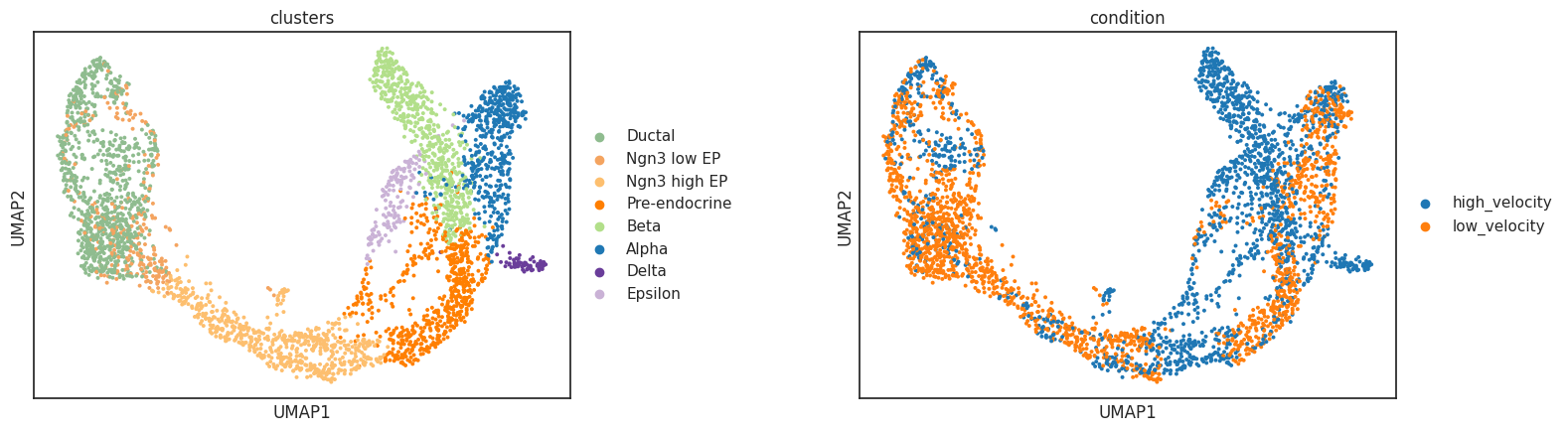
sc.pl.umap(adata, color=["condition", "clusters"], ncols=2, wspace=0.4,
title=["Artificial condition split", "Cell clusters"])
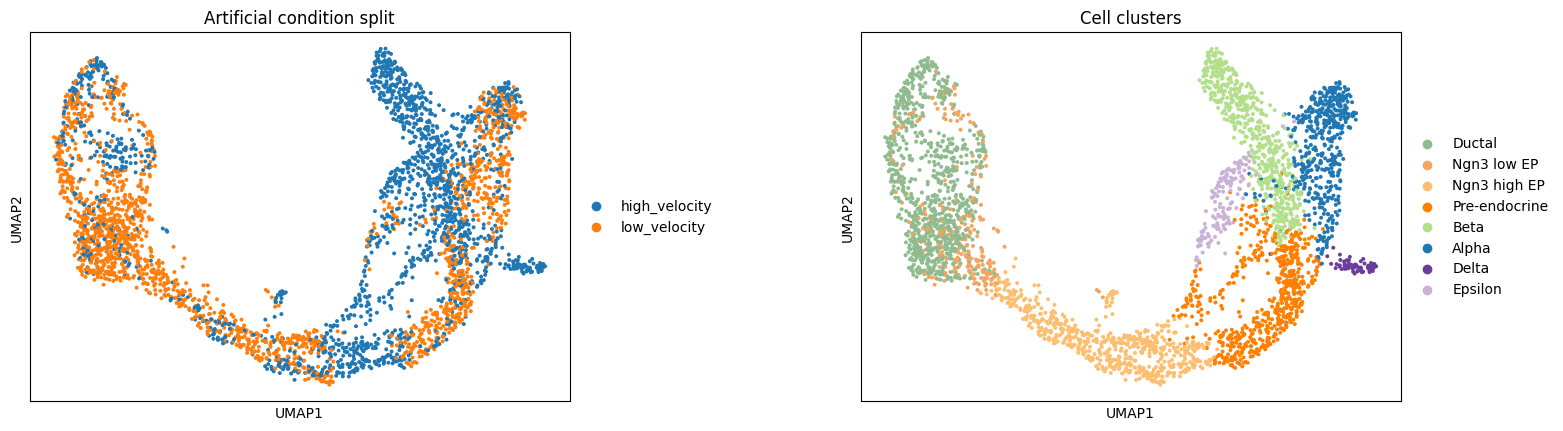
[6]:
# Confirm both conditions contain bifurcation + terminal fate cells
for cond in ["high_velocity", "low_velocity"]:
sub = adata[adata.obs["condition"] == cond]
clusters_present = sub.obs["clusters"].unique().tolist()
print(f"\n{cond}: {sub.n_obs} cells")
print(f" Clusters: {sorted(clusters_present)}")
high_velocity: 1848 cells
Clusters: ['Alpha', 'Beta', 'Delta', 'Ductal', 'Epsilon', 'Ngn3 high EP', 'Ngn3 low EP', 'Pre-endocrine']
low_velocity: 1848 cells
Clusters: ['Alpha', 'Beta', 'Ductal', 'Epsilon', 'Ngn3 high EP', 'Ngn3 low EP', 'Pre-endocrine']
3. Initialize PairScorer¶
PairScorer wraps SingleScorer and builds a shared embedding on pooled data from all conditions. This ensures that arm geometry is identical across conditions — a prerequisite for valid cross-condition comparison.
Key parameters:
condition_obs_key— column inadata.obswith condition labelsAll other parameters are identical to
SingleScorer
[7]:
mscorer = scCS.PairScorer(
adata,
root="Pre-endocrine",
branches=["Alpha", "Beta", "Delta", "Epsilon"],
condition_obs_key="condition", # our artificial split
obs_key="clusters",
n_angle_bins=36,
sector_method="centroid",
)
[scCS] PairScorer initialized.
Conditions (2): ['high_velocity', 'low_velocity']
Root: 'Pre-endocrine', Branches: ['Alpha', 'Beta', 'Delta', 'Epsilon']
Tier 1: Score each condition on the shared embedding¶
score_all_conditions() computes commitment scores separately for each condition, using the shared embedding and FateMap. Each condition’s cells are masked from the shared adata_sub.
[10]:
# Score all conditions
results = mscorer.score_all_conditions(
cell_level=True,
k_nn=15,
n_bootstrap=200, # bootstrap CI per condition
bootstrap_ci=0.95,
verbose=True,
)
[scCS] Scoring condition: 'high_velocity' (1249 cells)...
[scCS] Computing bootstrap CI (n=200)...
=== CommitmentScoreResult ===
Fates (4): Alpha, Beta, Delta, Epsilon
Dominant fate: Beta
Entropy metrics:
Population entropy: 0.6209 [aggregate velocity-mass balance]
Mean cell entropy: 0.9361 [per-cell average, k-way]
Per-fate cell entropy:
Alpha: 0.8246
Beta: 0.8608
Delta: 0.7541
Epsilon: 0.6399
NN-smoothed entropy (k=15): mean=0.9547 [per-cell, stored in adata_sub.obs['cs_nn_entropy']]
Commitment vector (normalized):
Alpha: 0.1490
Beta: 0.7267
Delta: 0.0663
Epsilon: 0.0580
Pairwise nCS matrix:
Alpha Beta Delta Epsilon
Alpha 1.000000 0.539098 0.756585 1.443795
Beta 1.854951 1.000000 1.403429 2.678169
Delta 1.321728 0.712540 1.000000 1.908304
Epsilon 0.692619 0.373389 0.524026 1.000000
Bootstrap 95% CI on nCS (n=200):
CI low:
Alpha Beta Delta Epsilon
Alpha 1.000 0.462 0.589 1.118
Beta 1.616 1.000 1.175 2.170
Delta 1.021 0.581 1.000 1.422
Epsilon 0.528 0.293 0.401 1.000
CI high:
Alpha Beta Delta Epsilon
Alpha 1.000 0.619 0.979 1.895
Beta 2.163 1.000 1.722 3.416
Delta 1.699 0.851 1.000 2.492
Epsilon 0.894 0.461 0.703 1.000
[scCS] Scoring condition: 'low_velocity' (627 cells)...
[scCS] Computing bootstrap CI (n=200)...
=== CommitmentScoreResult ===
Fates (4): Alpha, Beta, Delta, Epsilon
Dominant fate: Alpha
Entropy metrics:
Population entropy: 0.8152 [aggregate velocity-mass balance]
Mean cell entropy: 0.9361 [per-cell average, k-way]
Per-fate cell entropy:
Alpha: 0.8246
Beta: 0.8608
Delta: 0.7541
Epsilon: 0.6399
NN-smoothed entropy (k=15): mean=0.9547 [per-cell, stored in adata_sub.obs['cs_nn_entropy']]
Commitment vector (normalized):
Alpha: 0.4481
Beta: 0.3870
Delta: 0.1087
Epsilon: 0.0562
Pairwise nCS matrix:
Alpha Beta Delta Epsilon
Alpha 1.000000 0.186620 0.0 0.730455
Beta 5.358472 1.000000 0.0 3.914121
Delta inf inf 1.0 inf
Epsilon 1.369010 0.255485 0.0 1.000000
(inf = fate arm has 0 cells in this subset; expected for progenitor-only subsets)
Bootstrap 95% CI on nCS (n=200):
CI low:
Alpha Beta Delta Epsilon
Alpha 1.000 0.150 0.0 0.530
Beta 4.189 1.000 0.0 2.757
Delta NaN NaN 1.0 NaN
Epsilon 0.916 0.168 0.0 1.000
CI high:
Alpha Beta Delta Epsilon
Alpha 1.000 0.239 0.0 1.092
Beta 6.677 1.000 0.0 5.940
Delta NaN NaN 1.0 NaN
Epsilon 1.887 0.363 0.0 1.000
[11]:
# Compare nCS matrices side by side
for cond, res in results.items():
print(f"\n=== {cond} ===")
print(pd.DataFrame(
res.pairwise_nCS,
index=res.fate_names,
columns=res.fate_names
).round(3))
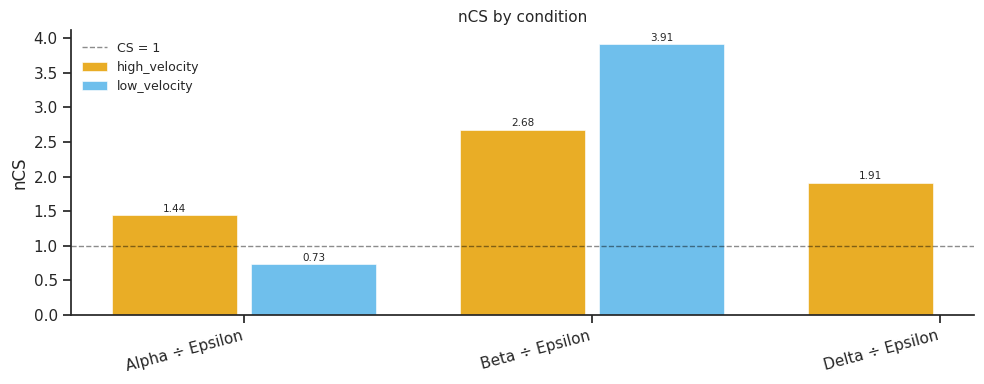
=== high_velocity ===
Alpha Beta Delta Epsilon
Alpha 1.000 0.539 0.757 1.444
Beta 1.855 1.000 1.403 2.678
Delta 1.322 0.713 1.000 1.908
Epsilon 0.693 0.373 0.524 1.000
=== low_velocity ===
Alpha Beta Delta Epsilon
Alpha 1.000 0.187 0.0 0.730
Beta 5.358 1.000 0.0 3.914
Delta inf inf 1.0 inf
Epsilon 1.369 0.255 0.0 1.000
[12]:
# Side-by-side star plots for each condition
# figsize_per_panel controls the size of each individual panel
fig = mscorer.plot_star_grid(
results,
figsize_per_panel=(7, 7),
)
plt.tight_layout()
plt.show()
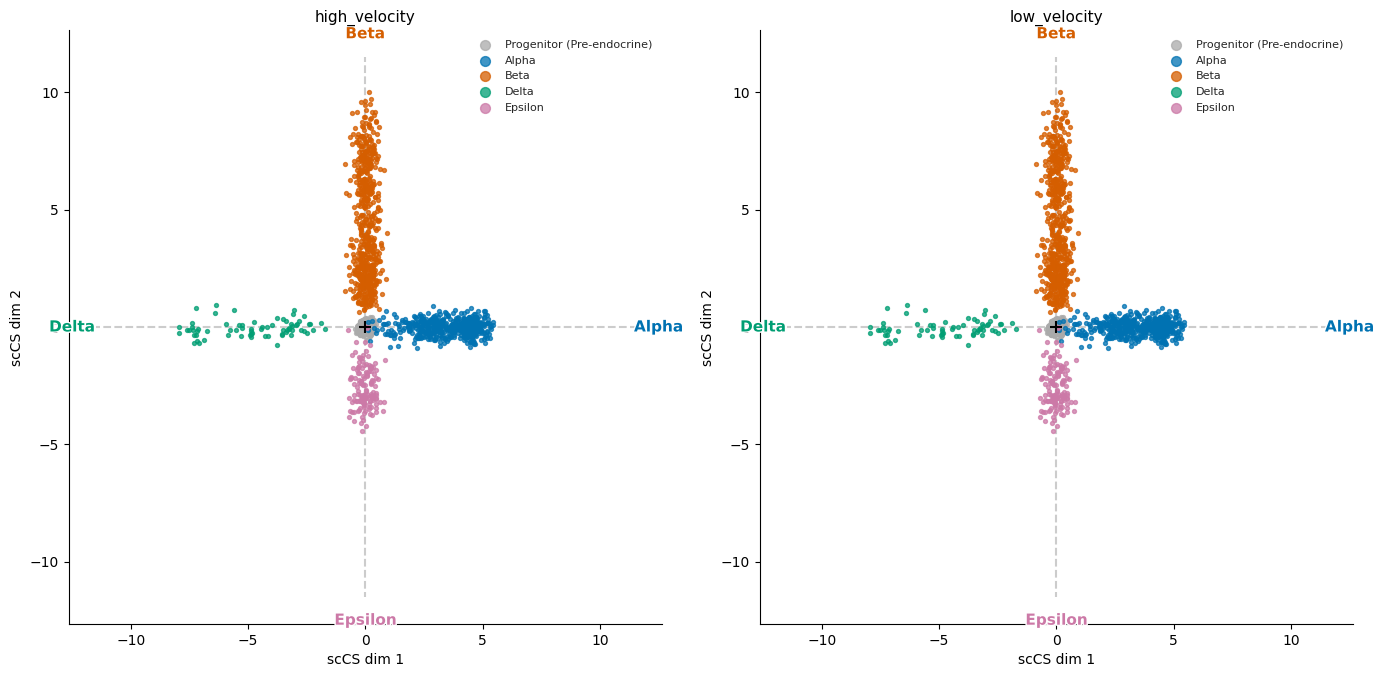
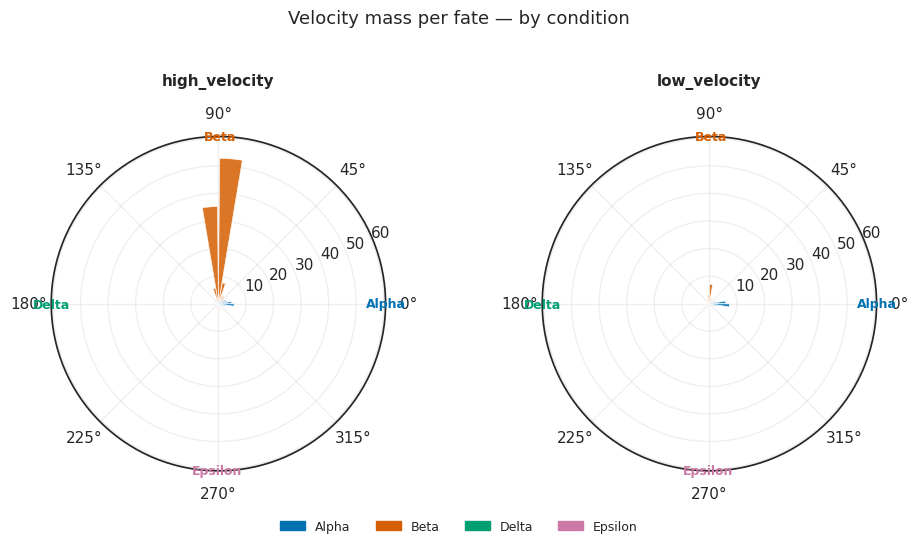
Tier 1b: Per-condition rose plots¶
plot_rose_grid() shows one polar rose plot per condition, all panels sharing the same radial scale for direct magnitude comparison across conditions.
[13]:
# Per-condition rose plots (shared radial scale)
fig = mscorer.plot_rose_grid(
results,
figsize_per_panel=(5, 5),
title="Velocity mass per fate — by condition",
)
plt.tight_layout()
plt.show()
[14]:
# Commitment vectors per condition
print("Commitment vectors (fraction of velocity mass per fate):")
for cond, res in results.items():
print(f"\n {cond}:")
for fate, cv in zip(res.fate_names, res.commitment_vector):
print(f" {fate}: {cv:.3f}")
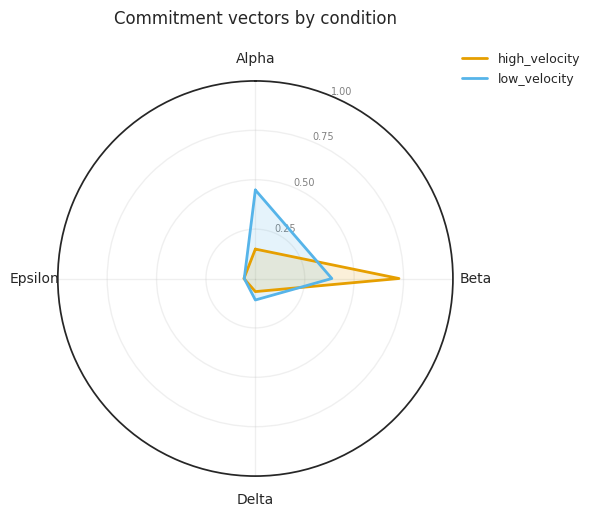
Commitment vectors (fraction of velocity mass per fate):
high_velocity:
Alpha: 0.149
Beta: 0.727
Delta: 0.066
Epsilon: 0.058
low_velocity:
Alpha: 0.448
Beta: 0.387
Delta: 0.109
Epsilon: 0.056
Tier 2: Statistical comparison¶
2a. ΔCS with bootstrap confidence intervals¶
compute_delta_CS() computes ΔnCS = nCS_A − nCS_B for each fate pair, with bootstrap CIs obtained by resampling cells within each condition.
A CI that does not include 0 indicates a statistically meaningful difference.
[15]:
# Compute ΔCS: high_velocity − low_velocity
delta = mscorer.compute_delta_CS(
condition_a="high_velocity",
condition_b="low_velocity",
n_bootstrap=500,
ci=0.95,
verbose=True,
)
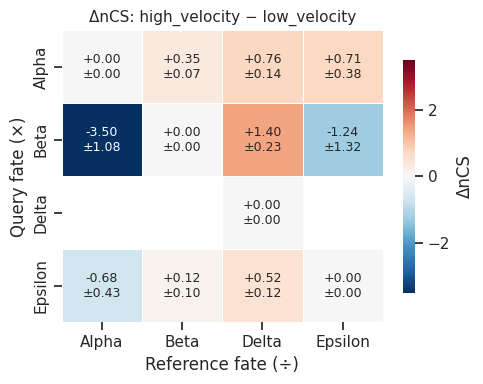
=== ΔCS: 'high_velocity' − 'low_velocity' ===
ΔnCS (point estimate):
Alpha Beta Delta Epsilon
Alpha 0.000 0.352 0.757 0.713
Beta -3.504 0.000 1.403 -1.236
Delta -inf -inf 0.000 -inf
Epsilon -0.676 0.118 0.524 0.000
95% CI low:
Alpha Beta Delta Epsilon
Alpha 0.000 0.279 0.640 0.317
Beta -4.659 0.000 1.210 -2.743
Delta NaN NaN 0.000 NaN
Epsilon -1.115 0.013 0.422 0.000
95% CI high:
Alpha Beta Delta Epsilon
Alpha 0.000 0.425 0.921 1.070
Beta -2.506 0.000 1.667 -0.106
Delta NaN NaN 0.000 NaN
Epsilon -0.259 0.209 0.654 0.000
[16]:
# Visualize ΔCS as a heatmap
import matplotlib.pyplot as plt
import matplotlib.colors as mcolors
fate_names = delta["fate_names"]
k = len(fate_names)
delta_mat = delta["delta_nCS"]
ci_low = delta["ci_low"]
ci_high = delta["ci_high"]
fig, axes = plt.subplots(1, 3, figsize=(14, 4))
for ax, mat, title in zip(
axes,
[delta_mat, ci_low, ci_high],
["ΔnCS (point estimate)", "95% CI — lower bound", "95% CI — upper bound"]
):
# Mask diagonal
masked = np.where(np.eye(k, dtype=bool), np.nan, mat)
vmax = np.nanmax(np.abs(masked))
im = ax.imshow(masked, cmap="RdBu_r", vmin=-vmax, vmax=vmax, aspect="auto")
ax.set_xticks(range(k)); ax.set_xticklabels(fate_names, rotation=45, ha="right")
ax.set_yticks(range(k)); ax.set_yticklabels(fate_names)
ax.set_title(title)
plt.colorbar(im, ax=ax, shrink=0.8)
plt.suptitle("ΔnCS: high_velocity − low_velocity", fontsize=13, y=1.02)
plt.tight_layout()
plt.show()
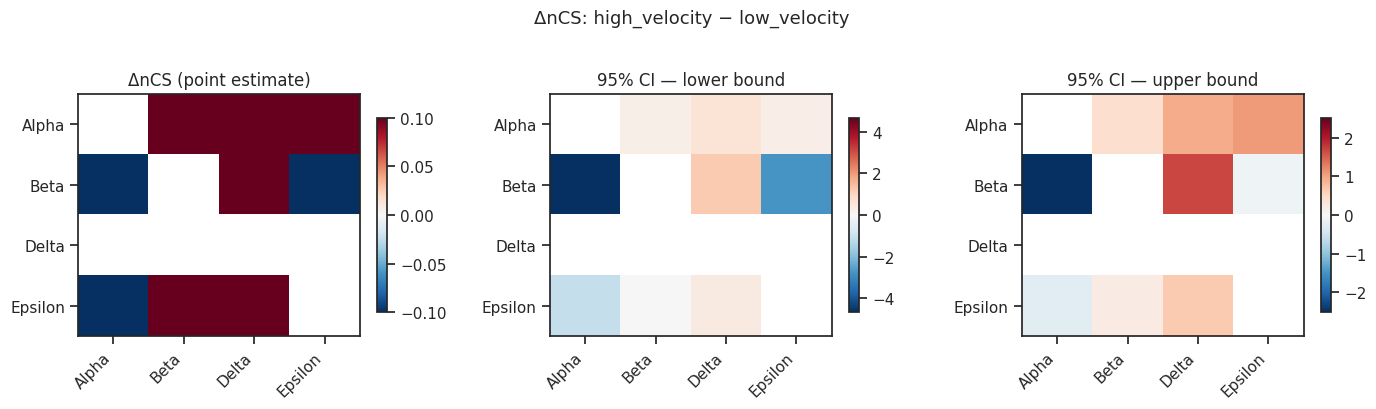
[17]:
# Identify fate pairs where CI excludes 0 (significant differences)
print("Fate pairs with significant ΔnCS (95% CI excludes 0):")
for i, fi in enumerate(fate_names):
for j, fj in enumerate(fate_names):
if i >= j:
continue
lo, hi = ci_low[i, j], ci_high[i, j]
if lo > 0 or hi < 0:
direction = "↑" if delta_mat[i, j] > 0 else "↓"
print(f" {fi} vs {fj}: Δ={delta_mat[i,j]:.3f} [{lo:.3f}, {hi:.3f}] {direction}")
Fate pairs with significant ΔnCS (95% CI excludes 0):
Alpha vs Beta: Δ=0.352 [0.279, 0.425] ↑
Alpha vs Delta: Δ=0.757 [0.640, 0.921] ↑
Alpha vs Epsilon: Δ=0.713 [0.317, 1.070] ↑
Beta vs Delta: Δ=1.403 [1.210, 1.667] ↑
Beta vs Epsilon: Δ=-1.236 [-2.743, -0.106] ↓
2b. Permutation test on per-cell fate affinities¶
compare_conditions() tests whether per-cell fate affinity scores differ significantly between conditions.
k=2 conditions: permutation test (shuffle condition labels, recompute mean difference)
k>2 conditions: Kruskal-Wallis + pairwise Mann-Whitney U with Bonferroni correction
[18]:
# Statistical comparison of per-cell fate affinities
stats_df = mscorer.compare_conditions(
results,
test="auto", # 'permutation' for k=2, 'kruskal' for k>2
n_permutations=1000,
pval_threshold=0.05,
verbose=True,
)
print("\nStatistical comparison results:")
print(stats_df.to_string(index=False))
=== Condition comparison ===
Test: auto | Significant results: 0 / 4
No significant differences at pval_adj < 0.05.
Statistical comparison results:
fate comparison test statistic pval mean_A mean_B pval_adj significant
Alpha high_velocity vs low_velocity kruskal-wallis 0.0 1.0 0.270815 0.270815 1.0 False
Beta high_velocity vs low_velocity kruskal-wallis 0.0 1.0 0.312566 0.312566 1.0 False
Delta high_velocity vs low_velocity kruskal-wallis 0.0 1.0 0.228806 0.228806 1.0 False
Epsilon high_velocity vs low_velocity kruskal-wallis 0.0 1.0 0.187813 0.187813 1.0 False
[19]:
# Violin / box / strip plots of per-cell fate affinities
fig = mscorer.plot_affinity_distributions(
results,
plot_type="violin", # 'violin', 'box', or 'strip'
figsize=(12, 5),
)
plt.tight_layout()
plt.show()
[20]:
# Box plot version
fig = mscorer.plot_affinity_distributions(
results,
plot_type="box",
figsize=(12, 5),
)
plt.tight_layout()
plt.show()
2c. Additional multi-condition visualizations¶
Three new plots for comparing commitment across conditions:
``plot_delta_cs_heatmap()`` — diverging heatmap of ΔnCS annotated with Δ ± CI
``plot_compare_conditions_bar()`` — grouped bar chart of nCS per condition per fate pair
``plot_commitment_vector_radar()`` — radar/spider chart of commitment vectors
[21]:
# ΔCS heatmap with CI annotation
fig = mscorer.plot_delta_cs_heatmap(
delta,
title="ΔnCS: high_velocity − low_velocity",
figsize=(5, 4),
)
plt.tight_layout()
plt.show()
[22]:
# Grouped bar chart: nCS per condition per fate pair
fig = mscorer.plot_compare_conditions_bar(
results,
title="nCS by condition",
figsize=(10, 4),
)
plt.tight_layout()
plt.show()
[23]:
# Radar chart of commitment vectors (one polygon per condition)
# Falls back to bar chart for k < 3
fig = mscorer.plot_commitment_vector_radar(
results,
title="Commitment vectors by condition",
figsize=(6, 6),
)
plt.tight_layout()
plt.show()
Tier 3: Trajectory-level analysis¶
3a. Mixed linear model¶
fit_mixed_model() fits a linear mixed model (via statsmodels MixedLM) for each fate arm:
affinity ~ condition (fixed effect)
+ (1 | sample) (random effect — accounts for replicate variation)
This is the most rigorous approach when you have biological replicates.
[24]:
# Fit mixed model: condition fixed effect, sample random effect
try:
lmm_results = mscorer.fit_mixed_model(
results,
replicate_key="sample", # replicate column
verbose=True,
)
print("\nMixed model results:")
print(lmm_results.to_string(index=False))
except Exception as e:
print(f"Mixed model skipped (requires statsmodels): {e}")
=== Mixed-effects model results ===
Significant effects: 0 / 4
Mixed model results:
fate condition reference coef std_err z_score pval ci_low ci_high pval_adj significant
Alpha low_velocity high_velocity -3.254918e-19 0.002280 -1.427620e-16 1.0 -0.004469 0.004469 1.0 False
Beta low_velocity high_velocity -1.775410e-18 0.003320 -5.346931e-16 1.0 -0.006508 0.006508 1.0 False
Delta low_velocity high_velocity -1.220594e-18 0.002279 -5.354918e-16 1.0 -0.004468 0.004468 1.0 False
Epsilon low_velocity high_velocity 3.030033e-16 0.003302 9.177422e-14 1.0 -0.006471 0.006471 1.0 False
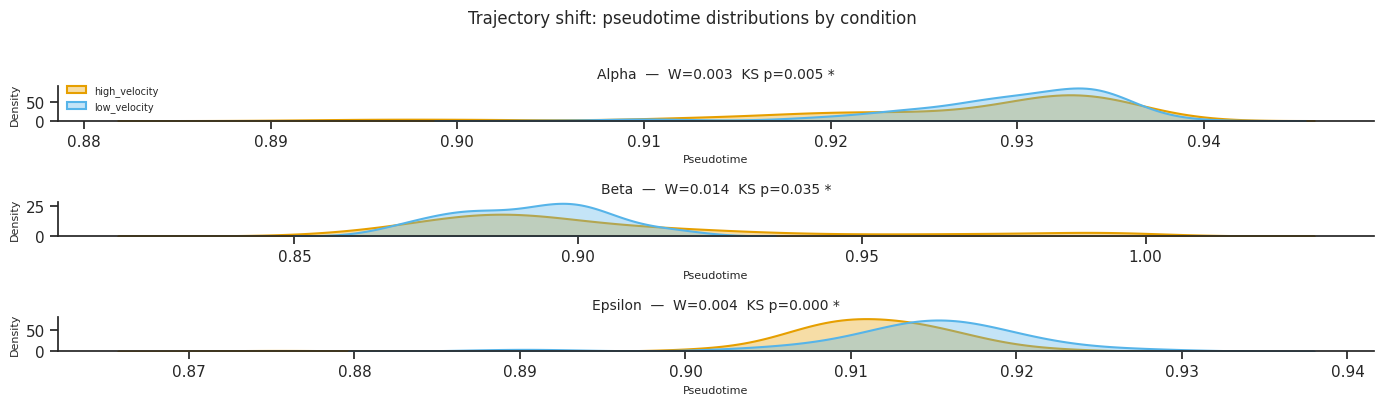
3b. Trajectory shift analysis¶
trajectory_shift() tests whether the pseudotime distribution along each fate arm differs between conditions using:
Kolmogorov-Smirnov (KS) test — detects any distributional difference
Wasserstein distance — quantifies the magnitude of the shift, with bootstrap CI
A significant KS test + large Wasserstein distance indicates that cells in one condition are systematically further along (or earlier in) the differentiation trajectory.
[25]:
# Trajectory shift analysis
# Returns a DataFrame with columns: fate, comparison, ks_stat, ks_pval,
# wasserstein, wasserstein_ci_low, wasserstein_ci_high, mean_pt_A, mean_pt_B,
# delta_mean_pt, significant
try:
shift_df = mscorer.trajectory_shift(
results,
n_bootstrap=500,
verbose=True,
)
print("\nTrajectory shift summary:")
print(shift_df[["fate", "comparison", "ks_stat", "ks_pval",
"wasserstein", "wasserstein_ci_low", "wasserstein_ci_high",
"significant"]].to_string(index=False))
except Exception as e:
print(f"Trajectory shift skipped: {e}")
shift_df = None
=== Trajectory shift analysis ===
Significant shifts: 3 / 3
fate comparison ks_stat ks_pval_adj wasserstein delta_mean_pt
Alpha high_velocity vs low_velocity 0.171245 0.005030 0.002727 -0.002664
Beta high_velocity vs low_velocity 0.246261 0.034612 0.014225 0.012765
Epsilon high_velocity vs low_velocity 0.481026 0.000210 0.003927 -0.003627
Trajectory shift summary:
fate comparison ks_stat ks_pval wasserstein wasserstein_ci_low wasserstein_ci_high significant
Alpha high_velocity vs low_velocity 0.171245 0.001677 0.002727 0.001802 0.004006 True
Beta high_velocity vs low_velocity 0.246261 0.011537 0.014225 0.012215 0.018473 True
Epsilon high_velocity vs low_velocity 0.481026 0.000070 0.003927 0.002510 0.006638 True
[26]:
# KDE plots of pseudotime distributions per fate per condition
try:
if shift_df is not None:
fig = mscorer.plot_trajectory_shift(
shift_df,
figsize=(14, 4),
)
plt.tight_layout()
plt.show()
except Exception as e:
print(f"Trajectory shift plot skipped: {e}")
6. Driver genes per condition¶
Identify fate-driving genes (velocity-based, DEG-based, velocity-fate correlation) within each condition. Use the shared adata_sub per condition, then assemble the results into one tidy table with (condition, fate) columns.
This is the standard way to find genes whose role in commitment changes between conditions: a gene that is a top driver in low_velocity but not in high_velocity is a candidate condition-specific regulator.
[27]:
# Velocity-based drivers — run per condition
from scCS.drivers import get_velocity_drivers
adata_sub_shared = mscorer._scorer.adata_sub
fate_names = mscorer._scorer._fate_map.fate_names
root_cluster = mscorer._scorer.root
obs_key_used = mscorer._scorer.obs_key
vel_drivers_per_cond = {}
for cond in mscorer.conditions:
mask = (adata_sub_shared.obs[mscorer.condition_obs_key].astype(str) == cond).values
sub = adata_sub_shared[mask].copy()
vel_drivers_per_cond[cond] = get_velocity_drivers(
sub, fate_names=fate_names, obs_key=obs_key_used,
root=root_cluster, n_top_genes=20,
)
# Compact long-form table
print("Top velocity drivers (top 5 per fate, per condition):")
_stack_drivers_by_condition(
vel_drivers_per_cond, n_per_fate=5,
cols=["gene", "mean_velocity", "rank"],
)
── Velocity drivers: Alpha (top 20, sorted by delta_velocity) ──
rank gene delta_velocity mean_velocity
1 Clu 2.394982 2.601254
2 Ghrl 2.230801 1.208408
3 Btbd17 1.361021 1.130396
4 Cryba2 1.165649 1.429237
5 Cpa2 0.996341 0.808371
6 Tm4sf4 0.752253 0.572645
7 Npepl1 0.701914 0.203834
8 Hn1 0.642953 0.173084
9 Pax6 0.628259 0.938433
10 Meis2 0.613610 0.691767
11 Vasp 0.563599 0.066359
12 Lurap1l 0.551248 0.355268
13 Rgs17 0.542472 0.575468
14 Celf3 0.540257 0.375474
15 Tox3 0.529739 0.308918
16 Cd200 0.518199 0.406608
17 Ppp3ca 0.514993 0.219860
18 Pax4 0.478970 0.058572
19 Cpt2 0.421590 0.253768
20 Smarcd2 0.420897 0.419561
── Velocity drivers: Beta (top 20, sorted by delta_velocity) ──
rank gene delta_velocity mean_velocity
1 Isl1 4.461863 1.070405
2 Ins2 3.953642 3.972451
3 Nnat 3.673498 4.141401
4 Clu 1.607484 1.813756
5 1700086L19Rik 1.450399 0.549565
6 Krt7 0.955820 0.266415
7 Igfbpl1 0.847445 0.290844
8 Tm4sf4 0.816697 0.637089
9 Pax4 0.810044 0.389647
10 Ppp1r1a 0.809831 1.533254
11 Ghrl 0.779220 -0.243173
12 Dbn1 0.643636 0.332081
13 Smarcd2 0.605017 0.603681
14 Pax6 0.562075 0.872248
15 Cpa2 0.534046 0.346076
16 Cldn6 0.472029 -0.293498
17 Glud1 0.468660 0.732603
18 Lurap1l 0.468362 0.272382
19 Ccnd2 0.465604 0.394593
20 BC023829 0.456752 0.379688
── Velocity drivers: Delta (top 20, sorted by delta_velocity) ──
rank gene delta_velocity mean_velocity
1 Krt8 2.332098 1.407045
2 Cpa2 2.280671 2.092702
3 Maged2 1.876646 3.380898
4 Ldha 1.703658 1.581080
5 Cryba2 1.640011 1.903599
6 Hn1 1.156748 0.686879
7 Cdkn1c 1.062967 1.007896
8 Nnat 1.012159 1.480061
9 Ambp 0.976446 0.752210
10 Ppp1r1a 0.971532 1.694955
11 Gnas 0.935484 6.604826
12 Rpl12 0.699341 -0.154604
13 Pax6 0.698164 1.008338
14 Sparc 0.694246 0.649847
15 Krt18 0.678155 -0.206350
16 Cotl1 0.649170 0.579984
17 Celf3 0.638144 0.473361
18 BC023829 0.593681 0.516617
19 Akr1c19 0.585637 1.123999
20 Foxa3 0.581431 0.411777
── Velocity drivers: Epsilon (top 20, sorted by delta_velocity) ──
rank gene delta_velocity mean_velocity
1 Cpa2 1.433100 1.245131
2 Cryba2 1.283010 1.546598
3 Clu 1.193620 1.399892
4 Meis2 0.974752 1.052909
5 Siva1 0.751692 0.628769
6 Btbd17 0.723855 0.493231
7 Lurap1l 0.644486 0.448505
8 Pax4 0.639611 0.219213
9 Syt13 0.626162 0.824190
10 Cdkn1c 0.600463 0.545391
11 Pax6 0.599071 0.909244
12 Rgs17 0.554066 0.587061
13 Nudt19 0.540254 0.460169
14 Arl6ip1 0.493820 -0.131398
15 Cpt2 0.455947 0.288125
16 Npepl1 0.448234 -0.049846
17 Birc5 0.410141 0.374991
18 Cdk1 0.397182 0.417446
19 Tkt 0.378253 0.332503
20 Ppp1r14b 0.371528 -0.291778
── Velocity drivers: Alpha (top 20, sorted by delta_velocity) ──
rank gene delta_velocity mean_velocity
1 Cpa2 1.493780 0.883649
2 Meis2 1.053937 0.645838
3 Cryba2 1.008127 1.055581
4 Pax6 0.848950 0.841338
5 Clu 0.848731 0.717436
6 Npepl1 0.757560 0.166575
7 Pax4 0.744543 0.016807
8 Krt7 0.716767 -0.259284
9 Vasp 0.652932 0.121307
10 Pdx1 0.634857 0.383033
11 Tm4sf4 0.611374 0.384856
12 Cd200 0.577635 0.271759
13 Celf3 0.529539 0.366640
14 BC023829 0.514373 0.350243
15 Tox3 0.480100 0.207980
16 Ppp3ca 0.463614 0.186181
17 Map1b 0.459361 0.640919
18 Krt8 0.457733 -0.503180
19 Psmc2 0.454385 -0.456419
20 Cpt2 0.447037 0.240750
── Velocity drivers: Beta (top 20, sorted by delta_velocity) ──
rank gene delta_velocity mean_velocity
1 Isl1 2.591787 1.067878
2 Nnat 2.565379 2.934238
3 Krt7 1.673100 0.697048
4 Meis2 1.406825 0.998726
5 Hadh 1.254540 1.095723
6 Ppp1r1a 0.992641 1.621375
7 Tm4sf4 0.900606 0.674089
8 BC023829 0.751555 0.587425
9 Pax6 0.649637 0.642025
10 Cpa2 0.648636 0.038504
11 Spc24 0.624380 0.625219
12 Lurap1l 0.589024 0.443184
13 Glud1 0.556597 0.805027
14 S100a10 0.545249 0.614486
15 Dlk1 0.522090 1.872635
16 Phgdh 0.512333 0.456298
17 Syt13 0.506459 0.347345
18 Map1b 0.486952 0.668510
19 Peg10 0.480236 0.988289
20 Gars 0.461482 0.495746
── Velocity drivers: Epsilon (top 20, sorted by delta_velocity) ──
rank gene delta_velocity mean_velocity
1 Isl1 1.954779 0.430871
2 Meis2 1.466027 1.057928
3 Pax4 1.266948 0.539212
4 Clu 1.176291 1.044996
5 Cryba2 1.061596 1.109050
6 Arl6ip1 1.010438 0.328101
7 Syt13 0.989686 0.830572
8 Pdx1 0.905458 0.653633
9 Pax6 0.798171 0.790560
10 Lurap1l 0.786681 0.640841
11 Ghrl 0.706006 0.556429
12 Runx1t1 0.666353 0.454053
13 Siva1 0.663069 0.555349
14 Cldn7 0.652394 -0.025911
15 Tkt 0.628411 0.701861
16 Nudt19 0.597660 0.495606
17 Npepl1 0.578077 -0.012909
18 Gars 0.554945 0.589210
19 Akr1c19 0.540149 0.622992
20 Nrep 0.521967 0.357101
Top velocity drivers (top 5 per fate, per condition):
[27]:
| condition | fate | gene | mean_velocity | rank | |
|---|---|---|---|---|---|
| 0 | high_velocity | Alpha | Clu | 2.601254 | 1 |
| 1 | high_velocity | Alpha | Ghrl | 1.208408 | 2 |
| 2 | high_velocity | Alpha | Btbd17 | 1.130396 | 3 |
| 3 | high_velocity | Alpha | Cryba2 | 1.429237 | 4 |
| 4 | high_velocity | Alpha | Cpa2 | 0.808371 | 5 |
| ... | ... | ... | ... | ... | ... |
| 30 | low_velocity | Epsilon | Isl1 | 0.430871 | 1 |
| 31 | low_velocity | Epsilon | Meis2 | 1.057928 | 2 |
| 32 | low_velocity | Epsilon | Pax4 | 0.539212 | 3 |
| 33 | low_velocity | Epsilon | Clu | 1.044996 | 4 |
| 34 | low_velocity | Epsilon | Cryba2 | 1.109050 | 5 |
35 rows × 5 columns
[28]:
# DEG-based drivers — run per condition
from scCS.drivers import get_deg_drivers
deg_drivers_per_cond = {}
for cond in mscorer.conditions:
mask = (adata_sub_shared.obs[mscorer.condition_obs_key].astype(str) == cond).values
sub = adata_sub_shared[mask].copy()
deg_drivers_per_cond[cond] = get_deg_drivers(
sub, fate_names=fate_names, obs_key=obs_key_used,
root=root_cluster, n_top_genes=20,
pval_threshold=0.05, logfc_threshold=0.25,
)
print("Top DEG drivers (top 5 per fate, per condition):")
_stack_drivers_by_condition(
deg_drivers_per_cond, n_per_fate=5,
cols=["gene", "logfoldchange", "pval_adj", "pct_fate", "pct_progenitor"],
)
── DEG drivers: Alpha vs progenitor ──
Significant: 531 (up: 251, down: 280)
gene logfoldchange pval_adj pct_fate pct_progenitor
Gcg 270.465210 1.082532e-38 0.735577 NaN
Pyy 168.419342 2.504105e-31 0.961538 NaN
Iapp 103.014679 1.340236e-46 0.855769 NaN
Ttr 72.689537 6.161996e-51 1.000000 NaN
Gnas 36.008827 8.249209e-28 1.000000 NaN
Rbp4 32.906731 1.717601e-17 0.942308 NaN
Slc38a5 19.159832 4.085392e-32 0.903846 NaN
Tmem27 15.835037 5.708899e-60 0.971154 NaN
Pcsk1n 14.708415 1.348048e-36 0.995192 NaN
Gast 10.758981 8.631471e-15 0.548077 NaN
Cpe 8.463097 1.488365e-23 1.000000 NaN
Pcsk2 7.870811 5.754125e-33 0.841346 NaN
Gpx3 7.583188 3.834793e-39 0.937500 NaN
Ppy 6.445889 2.303623e-07 0.408654 NaN
Ppp1r1a 5.905676 1.546018e-17 0.480769 NaN
Peg10 5.882595 7.186934e-31 0.745192 NaN
Meis2 5.551431 8.249209e-28 0.966346 NaN
Tmsb15l 5.335431 5.589635e-34 0.725962 NaN
Ssr2 5.193390 3.089435e-23 0.975962 NaN
Slc30a8 5.180974 3.695947e-13 0.423077 NaN
── DEG drivers: Beta vs progenitor ──
Significant: 701 (up: 357, down: 344)
gene logfoldchange pval_adj pct_fate pct_progenitor
Iapp 198.310974 6.535262e-104 0.954296 NaN
Pyy 113.021301 1.023042e-36 0.963437 NaN
Ins1 106.606689 1.386384e-33 0.608775 NaN
Ins2 50.743568 1.352767e-49 0.647166 NaN
Nnat 45.320721 3.310466e-74 0.848263 NaN
Rbp4 34.660763 4.577475e-51 0.998172 NaN
Gnas 31.077551 9.071092e-51 1.000000 NaN
P2ry1 27.969667 7.949997e-05 0.177331 NaN
Bace2 27.218302 2.121908e-02 0.111517 NaN
Ttr 23.233101 3.446132e-37 0.994516 NaN
Sec61b 13.315624 3.730541e-86 1.000000 NaN
Dlk1 12.743445 8.416356e-64 0.976234 NaN
Npy 12.622334 4.217475e-06 0.206581 NaN
Ppp1r1a 12.056191 5.953017e-69 0.744059 NaN
Pcsk2 9.823764 1.333376e-91 0.990859 NaN
Calr 8.645715 5.277221e-60 0.967093 NaN
Hspa5 8.439935 1.700786e-68 0.972578 NaN
Rpl10 8.116318 7.607530e-21 1.000000 NaN
Pcsk1n 7.766905 5.758600e-34 0.994516 NaN
G6pc2 7.745673 4.441791e-27 0.460695 NaN
── DEG drivers: Delta vs progenitor ──
Significant: 351 (up: 147, down: 204)
gene logfoldchange pval_adj pct_fate pct_progenitor
Pyy 458.501343 6.517585e-28 1.000000 NaN
Sst 330.755005 3.134982e-26 0.871429 NaN
Rbp4 187.659607 1.393899e-32 1.000000 NaN
Iapp 102.244209 9.906281e-32 0.985714 NaN
Ptprz1 28.902637 6.250994e-03 0.257143 NaN
Cd24a 18.501904 8.042192e-25 1.000000 NaN
Ppy 17.762150 2.337212e-05 0.485714 NaN
Dlk1 16.546801 4.103858e-24 1.000000 NaN
Hhex 14.889072 1.006595e-22 0.985714 NaN
Rpl10 12.829304 1.254427e-10 1.000000 NaN
Gpx3 9.945301 6.213992e-23 0.985714 NaN
Ssr2 9.680505 2.257681e-20 1.000000 NaN
Pcsk2 8.724628 2.377749e-20 0.928571 NaN
Ttr 8.637319 3.633579e-03 0.971429 NaN
Hadh 8.508456 7.194880e-25 0.971429 NaN
Arg1 7.800181 9.417072e-25 0.900000 NaN
Spock3 7.769428 1.303711e-04 0.342857 NaN
Mest 7.046804 6.562641e-16 0.714286 NaN
Igfbp7 6.177687 3.243405e-15 0.685714 NaN
Stk32a 6.167919 3.549981e-02 0.214286 NaN
── DEG drivers: Epsilon vs progenitor ──
Significant: 418 (up: 213, down: 205)
gene logfoldchange pval_adj pct_fate pct_progenitor
Ghrl 475.082031 2.203349e-52 1.000000 NaN
Rbp4 73.654526 1.484470e-27 0.991453 NaN
Pyy 63.465481 2.267171e-07 0.880342 NaN
Gcg 23.913074 6.375154e-05 0.393162 NaN
Iapp 20.790907 9.597633e-18 0.769231 NaN
Ttr 20.403343 3.696554e-04 0.914530 NaN
Lrpprc 14.690345 1.281113e-07 0.871795 NaN
Cdkn1a 12.742599 7.097317e-03 0.863248 NaN
Isl1 12.671727 1.202554e-12 0.991453 NaN
Maged2 11.401874 1.496087e-23 0.931624 NaN
Tmem27 9.584536 2.461019e-20 0.811966 NaN
Slc38a5 8.596473 4.696298e-10 0.854701 NaN
Hspa5 8.034565 7.664804e-23 0.948718 NaN
Ssr2 8.016420 1.174690e-27 0.991453 NaN
Calr 7.737453 2.579614e-24 0.948718 NaN
Rpl10 6.453340 1.565684e-05 1.000000 NaN
Ccnd2 5.899344 3.823843e-20 0.692308 NaN
Arg1 5.192980 2.499013e-18 0.683761 NaN
Pdia6 5.182480 6.791899e-21 0.888889 NaN
Acsl1 5.174711 3.383417e-03 0.230769 NaN
── DEG drivers: Alpha vs progenitor ──
Significant: 574 (up: 268, down: 306)
gene logfoldchange pval_adj pct_fate pct_progenitor
Pyy 249.362976 9.931852e-67 0.974359 NaN
Gcg 163.256683 1.304384e-26 0.597070 NaN
Iapp 96.747475 6.068537e-52 0.853480 NaN
Ttr 63.648888 1.081201e-50 1.000000 NaN
Rbp4 36.691395 3.598094e-36 0.978022 NaN
Gnas 32.365688 1.523612e-23 1.000000 NaN
Pou6f2 28.212460 3.469651e-03 0.164835 NaN
Tmem27 18.896784 1.606955e-79 0.989011 NaN
Slc38a5 12.228434 2.892680e-27 0.934066 NaN
Gast 10.213935 2.636313e-18 0.531136 NaN
Pcsk1n 9.659978 1.579846e-21 1.000000 NaN
Rpl10 7.830467 9.942312e-13 1.000000 NaN
Meis2 7.603310 2.573396e-50 0.974359 NaN
Peg10 7.555929 6.845952e-35 0.666667 NaN
Gpx3 7.432570 3.120716e-42 0.937729 NaN
Pcsk2 7.368222 1.115644e-28 0.849817 NaN
Ssr2 7.260166 2.593563e-46 0.992674 NaN
Dpp4 7.053434 2.538136e-11 0.347985 NaN
Isl1 6.900289 8.662467e-24 0.882784 NaN
Ripply3 6.896678 7.616812e-13 0.373626 NaN
── DEG drivers: Beta vs progenitor ──
Significant: 203 (up: 98, down: 105)
gene logfoldchange pval_adj pct_fate pct_progenitor
Pyy 172.267761 1.129461e-13 0.954545 NaN
Rbp4 42.132294 5.798260e-14 0.977273 NaN
Ttr 29.687593 2.551584e-07 1.000000 NaN
Gnas 22.671938 1.931650e-05 1.000000 NaN
Iapp 20.322002 2.575326e-10 0.818182 NaN
1700086L19Rik 9.403206 5.302802e-12 1.000000 NaN
Lrpprc 8.379879 9.063213e-07 0.977273 NaN
Pcsk1n 7.748258 5.745179e-05 1.000000 NaN
Rpl10 7.554930 1.799808e-02 1.000000 NaN
Sec61b 6.042389 2.968468e-07 1.000000 NaN
Pcsk2 5.976448 1.866927e-09 0.909091 NaN
Tmem27 5.387622 2.001496e-09 0.795455 NaN
Immp1l 5.075066 9.122668e-10 0.977273 NaN
Ociad2 5.014332 7.697577e-11 0.750000 NaN
Dlk1 4.804500 8.567286e-05 0.840909 NaN
Ssr2 4.574903 1.371557e-08 1.000000 NaN
Slc38a5 4.565075 8.569488e-03 0.795455 NaN
Isl1 4.369471 2.541336e-06 0.931818 NaN
Ppp1r1a 4.367425 2.167165e-02 0.318182 NaN
Nnat 4.217022 7.842166e-03 0.545455 NaN
── DEG drivers: Epsilon vs progenitor ──
Significant: 186 (up: 96, down: 90)
gene logfoldchange pval_adj pct_fate pct_progenitor
Ghrl 586.200867 2.348411e-13 1.00 NaN
Pyy 73.462746 2.327392e-05 0.84 NaN
Rbp4 54.519619 1.337757e-09 1.00 NaN
Lrpprc 48.062851 6.402959e-07 0.88 NaN
Cdkn1a 40.506321 1.098848e-12 1.00 NaN
Maged2 30.468407 1.053974e-12 1.00 NaN
Isl1 29.391127 1.125649e-12 1.00 NaN
Enpp1 28.794609 3.381167e-02 0.36 NaN
Arg1 15.583445 1.098848e-12 0.96 NaN
Hspa5 14.280921 4.614881e-11 1.00 NaN
Calr 11.404353 1.763643e-09 0.96 NaN
Gch1 9.612990 2.085134e-03 1.00 NaN
Fam183b 9.241714 5.915602e-08 1.00 NaN
Iapp 9.183792 2.282038e-03 0.72 NaN
Mboat4 8.834090 1.298473e-08 0.80 NaN
Acsl1 8.229982 6.131174e-03 0.44 NaN
Rpl10 8.060061 1.383082e-02 1.00 NaN
Peg3 7.796899 1.263498e-03 1.00 NaN
Irx2 7.542091 8.854235e-06 0.64 NaN
Serpina1c 7.235207 6.218541e-07 0.84 NaN
Top DEG drivers (top 5 per fate, per condition):
[28]:
| condition | fate | gene | logfoldchange | pval_adj | pct_fate | pct_progenitor | |
|---|---|---|---|---|---|---|---|
| 0 | high_velocity | Alpha | Gcg | 270.465210 | 1.082532e-38 | 0.735577 | NaN |
| 1 | high_velocity | Alpha | Pyy | 168.419342 | 2.504105e-31 | 0.961538 | NaN |
| 2 | high_velocity | Alpha | Iapp | 103.014679 | 1.340236e-46 | 0.855769 | NaN |
| 3 | high_velocity | Alpha | Ttr | 72.689537 | 6.161996e-51 | 1.000000 | NaN |
| 4 | high_velocity | Alpha | Gnas | 36.008827 | 8.249209e-28 | 1.000000 | NaN |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 30 | low_velocity | Epsilon | Ghrl | 586.200867 | 2.348411e-13 | 1.000000 | NaN |
| 31 | low_velocity | Epsilon | Pyy | 73.462746 | 2.327392e-05 | 0.840000 | NaN |
| 32 | low_velocity | Epsilon | Rbp4 | 54.519619 | 1.337757e-09 | 1.000000 | NaN |
| 33 | low_velocity | Epsilon | Lrpprc | 48.062851 | 6.402959e-07 | 0.880000 | NaN |
| 34 | low_velocity | Epsilon | Cdkn1a | 40.506321 | 1.098848e-12 | 1.000000 | NaN |
35 rows × 7 columns
[29]:
# Driver overlap between conditions: which genes are shared vs unique per fate?
def _driver_overlap(per_cond_drivers, n_top=10, gene_col="gene"):
"""Build a fate x gene x condition presence matrix from {cond: {fate: df}}.
Defensive: missing fates / missing gene columns / empty DataFrames yield empty sets.
"""
rows = []
conds = list(per_cond_drivers.keys())
if not conds:
return pd.DataFrame()
# Union of fates across all conditions (don't assume identical fate sets)
fates = sorted({f for d in per_cond_drivers.values() for f in d.keys()})
for fate in fates:
sets = {}
for cond in conds:
df = per_cond_drivers[cond].get(fate)
if df is None or len(df) == 0 or gene_col not in df.columns:
sets[cond] = set()
else:
sets[cond] = set(df.head(n_top)[gene_col].astype(str))
all_genes = sorted(set.union(*sets.values())) if sets else []
for g in all_genes:
row = {"fate": fate, "gene": g}
for cond in conds:
row[cond] = g in sets[cond]
row["n_conds"] = sum(row[cond] for cond in conds)
row["shared_all"] = row["n_conds"] == len(conds)
rows.append(row)
return pd.DataFrame(rows)
overlap_df = _driver_overlap(vel_drivers_per_cond, n_top=10)
print(f"Top-10 velocity driver overlap (per fate, {len(mscorer.conditions)} conditions):")
overlap_df.head(20)
Top-10 velocity driver overlap (per fate, 2 conditions):
[29]:
| fate | gene | high_velocity | low_velocity | n_conds | shared_all | |
|---|---|---|---|---|---|---|
| 0 | Alpha | Btbd17 | True | False | 1 | False |
| 1 | Alpha | Clu | True | True | 2 | True |
| 2 | Alpha | Cpa2 | True | True | 2 | True |
| 3 | Alpha | Cryba2 | True | True | 2 | True |
| 4 | Alpha | Ghrl | True | False | 1 | False |
| 5 | Alpha | Hn1 | True | False | 1 | False |
| 6 | Alpha | Krt7 | False | True | 1 | False |
| 7 | Alpha | Meis2 | True | True | 2 | True |
| 8 | Alpha | Npepl1 | True | True | 2 | True |
| 9 | Alpha | Pax4 | False | True | 1 | False |
| 10 | Alpha | Pax6 | True | True | 2 | True |
| 11 | Alpha | Pdx1 | False | True | 1 | False |
| 12 | Alpha | Tm4sf4 | True | False | 1 | False |
| 13 | Alpha | Vasp | False | True | 1 | False |
| 14 | Beta | 1700086L19Rik | True | False | 1 | False |
| 15 | Beta | BC023829 | False | True | 1 | False |
| 16 | Beta | Clu | True | False | 1 | False |
| 17 | Beta | Cpa2 | False | True | 1 | False |
| 18 | Beta | Hadh | False | True | 1 | False |
| 19 | Beta | Igfbpl1 | True | False | 1 | False |
7. Pathway enrichment per condition¶
Run get_enrichment per condition on the DEG drivers computed above. Display top pathways per (condition, fate, direction) in a single compact table.
Requires
gseapyand internet access for the Enrichr API. Wrapped in try/except so the notebook still builds offline.
[30]:
# Pathway enrichment — run per condition on the DEG drivers from §6
from scCS.enrichment import run_enrichment_per_fate
enrichment_per_cond = {}
for cond in mscorer.conditions:
try:
enrichment_per_cond[cond] = run_enrichment_per_fate(
deg_drivers=deg_drivers_per_cond[cond],
fate_names=fate_names,
organism="mouse",
pval_threshold=0.05,
logfc_threshold=0.25,
plot=False,
n_top_pathways=10,
)
except ImportError:
print("gseapy not installed. Run: pip install gseapy")
break
except Exception as e:
print(f"Enrichment for '{cond}' skipped: {e}")
enrichment_per_cond[cond] = {}
# Compact summary across conditions
enrich_df = _stack_enrichment_by_condition(enrichment_per_cond, n_per_group=3)
if not enrich_df.empty:
keep = ["condition", "fate", "direction", "Gene_set", "Term",
"Overlap", "Adjusted P-value"]
keep = [c for c in keep if c in enrich_df.columns]
display(enrich_df[keep])
else:
print("(No enriched terms — common with small datasets / few significant DEGs.)")
============================================================
Pathway enrichment: Alpha
Gene sets: ['KEGG_2019_Mouse', 'GO_Biological_Process_2021', 'Reactome_2022']
Up-regulated genes : 251
Down-regulated genes: 280
============================================================
[up] Significant terms: 61
Gene_set Term Overlap Adjusted P-value
KEGG_2019_Mouse Protein processing in endoplasmic reticulum 14/163 0.000004
KEGG_2019_Mouse Thyroid hormone synthesis 9/73 0.000032
KEGG_2019_Mouse Circadian entrainment 8/99 0.001974
KEGG_2019_Mouse Gastric acid secretion 7/74 0.001974
KEGG_2019_Mouse Lysosome 8/124 0.006893
GO_Biological_Process_2021 regulation of insulin secretion (GO:0050796) 9/104 0.010186
KEGG_2019_Mouse Dopaminergic synapse 8/135 0.010271
KEGG_2019_Mouse Protein export 4/28 0.011122
KEGG_2019_Mouse Retrograde endocannabinoid signaling 8/150 0.013541
KEGG_2019_Mouse Gap junction 6/86 0.013541
============================================================
Pathway enrichment: Beta
Gene sets: ['KEGG_2019_Mouse', 'GO_Biological_Process_2021', 'Reactome_2022']
Up-regulated genes : 357
Down-regulated genes: 344
============================================================
[up] Significant terms: 156
Gene_set Term Overlap Adjusted P-value
KEGG_2019_Mouse Protein processing in endoplasmic reticulum 22/163 4.044027e-11
GO_Biological_Process_2021 IRE1-mediated unfolded protein response (GO:0036498) 12/53 2.514449e-07
KEGG_2019_Mouse Insulin secretion 13/86 4.749228e-07
Reactome_2022 IRE1alpha Activates Chaperones R-HSA-381070 10/48 7.204516e-06
Reactome_2022 Unfolded Protein Response (UPR) R-HSA-381119 12/89 2.090815e-05
Reactome_2022 XBP1(S) Activates Chaperone Genes R-HSA-381038 9/46 2.371736e-05
KEGG_2019_Mouse Maturity onset diabetes of the young 7/27 2.784418e-05
KEGG_2019_Mouse Thyroid hormone synthesis 10/73 3.894807e-05
Reactome_2022 Integration Of Energy Metabolism R-HSA-163685 12/105 6.656112e-05
Reactome_2022 Hemostasis R-HSA-109582 29/576 8.070363e-05
[down] Significant terms: 134
Gene_set Term Overlap Adjusted P-value
GO_Biological_Process_2021 cotranslational protein targeting to membrane (GO:0006613) 27/94 8.616112e-23
GO_Biological_Process_2021 cytoplasmic translation (GO:0002181) 27/93 8.616112e-23
GO_Biological_Process_2021 SRP-dependent cotranslational protein targeting to membrane (GO:0006614) 26/90 4.000975e-22
Reactome_2022 Peptide Chain Elongation R-HSA-156902 25/86 3.483571e-21
Reactome_2022 Eukaryotic Translation Elongation R-HSA-156842 25/90 4.119235e-21
Reactome_2022 Eukaryotic Translation Termination R-HSA-72764 25/90 4.119235e-21
Reactome_2022 Nonsense Mediated Decay (NMD) Independent Of Exon Junction Complex (EJC) R-HSA-975956 25/92 5.670726e-21
GO_Biological_Process_2021 nuclear-transcribed mRNA catabolic process, nonsense-mediated decay (GO:0000184) 27/113 9.923864e-21
GO_Biological_Process_2021 protein targeting to ER (GO:0045047) 26/103 1.143783e-20
Reactome_2022 Viral mRNA Translation R-HSA-192823 24/90 4.789000e-20
============================================================
Pathway enrichment: Delta
Gene sets: ['KEGG_2019_Mouse', 'GO_Biological_Process_2021', 'Reactome_2022']
Up-regulated genes : 147
Down-regulated genes: 204
============================================================
[up] Significant terms: 39
Gene_set Term Overlap Adjusted P-value
KEGG_2019_Mouse Protein processing in endoplasmic reticulum 14/163 2.832562e-09
KEGG_2019_Mouse Thyroid hormone synthesis 7/73 9.105573e-05
KEGG_2019_Mouse Circadian entrainment 7/99 4.730875e-04
KEGG_2019_Mouse Protein export 4/28 2.087400e-03
KEGG_2019_Mouse Dopaminergic synapse 7/135 2.118750e-03
GO_Biological_Process_2021 IRE1-mediated unfolded protein response (GO:0036498) 6/53 2.941985e-03
KEGG_2019_Mouse Long-term potentiation 5/67 3.732861e-03
KEGG_2019_Mouse Cholinergic synapse 6/113 4.481356e-03
KEGG_2019_Mouse Gastric acid secretion 5/74 4.481356e-03
Reactome_2022 SRP-dependent Cotranslational Protein Targeting To Membrane R-HSA-1799339 7/108 4.707509e-03
============================================================
Pathway enrichment: Epsilon
Gene sets: ['KEGG_2019_Mouse', 'GO_Biological_Process_2021', 'Reactome_2022']
Up-regulated genes : 213
Down-regulated genes: 205
============================================================
[down] Significant terms: 398
Gene_set Term Overlap Adjusted P-value
GO_Biological_Process_2021 cotranslational protein targeting to membrane (GO:0006613) 26/94 1.730020e-27
GO_Biological_Process_2021 cytoplasmic translation (GO:0002181) 26/93 1.730020e-27
GO_Biological_Process_2021 SRP-dependent cotranslational protein targeting to membrane (GO:0006614) 25/90 1.400350e-26
GO_Biological_Process_2021 nuclear-transcribed mRNA catabolic process, nonsense-mediated decay (GO:0000184) 26/113 1.771113e-25
Reactome_2022 Peptide Chain Elongation R-HSA-156902 24/86 2.479010e-25
Reactome_2022 Eukaryotic Translation Elongation R-HSA-156842 24/90 2.829020e-25
Reactome_2022 Eukaryotic Translation Termination R-HSA-72764 24/90 2.829020e-25
GO_Biological_Process_2021 protein targeting to ER (GO:0045047) 25/103 3.642322e-25
Reactome_2022 Nonsense Mediated Decay (NMD) Independent Of Exon Junction Complex (EJC) R-HSA-975956 24/92 3.831354e-25
Reactome_2022 Selenocysteine Synthesis R-HSA-2408557 23/90 5.502352e-24
============================================================
Pathway enrichment: Alpha
Gene sets: ['KEGG_2019_Mouse', 'GO_Biological_Process_2021', 'Reactome_2022']
Up-regulated genes : 268
Down-regulated genes: 306
============================================================
[up] Significant terms: 69
Gene_set Term Overlap Adjusted P-value
KEGG_2019_Mouse Protein processing in endoplasmic reticulum 16/163 1.379291e-07
KEGG_2019_Mouse Thyroid hormone synthesis 9/73 5.845004e-05
KEGG_2019_Mouse Lysosome 10/124 4.694061e-04
KEGG_2019_Mouse Protein export 5/28 1.660431e-03
GO_Biological_Process_2021 regulation of insulin secretion (GO:0050796) 10/104 2.430781e-03
KEGG_2019_Mouse Gastric acid secretion 7/74 2.509346e-03
KEGG_2019_Mouse Glycosaminoglycan degradation 4/21 5.491478e-03
GO_Biological_Process_2021 IRE1-mediated unfolded protein response (GO:0036498) 7/53 5.826129e-03
Reactome_2022 Peptide Hormone Metabolism R-HSA-2980736 8/89 9.062283e-03
Reactome_2022 Asparagine N-linked Glycosylation R-HSA-446203 13/282 9.062283e-03
[down] Significant terms: 297
Gene_set Term Overlap Adjusted P-value
GO_Biological_Process_2021 cytoplasmic translation (GO:0002181) 23/93 1.520504e-18
GO_Biological_Process_2021 cotranslational protein targeting to membrane (GO:0006613) 23/94 1.520504e-18
Reactome_2022 Eukaryotic Translation Termination R-HSA-72764 22/90 3.639478e-18
Reactome_2022 Peptide Chain Elongation R-HSA-156902 22/86 3.639478e-18
Reactome_2022 Eukaryotic Translation Elongation R-HSA-156842 22/90 3.639478e-18
Reactome_2022 Nonsense Mediated Decay (NMD) Independent Of Exon Junction Complex (EJC) R-HSA-975956 22/92 4.603451e-18
GO_Biological_Process_2021 SRP-dependent cotranslational protein targeting to membrane (GO:0006614) 22/90 7.996281e-18
Reactome_2022 Viral mRNA Translation R-HSA-192823 21/90 4.007504e-17
Reactome_2022 Selenocysteine Synthesis R-HSA-2408557 21/90 4.007504e-17
GO_Biological_Process_2021 nuclear-transcribed mRNA catabolic process, nonsense-mediated decay (GO:0000184) 23/113 6.885432e-17
============================================================
Pathway enrichment: Beta
Gene sets: ['KEGG_2019_Mouse', 'GO_Biological_Process_2021', 'Reactome_2022']
Up-regulated genes : 98
Down-regulated genes: 105
============================================================
[up] Significant terms: 42
Gene_set Term Overlap Adjusted P-value
KEGG_2019_Mouse Protein processing in endoplasmic reticulum 10/163 0.000001
Reactome_2022 Response To Elevated Platelet Cytosolic Ca2+ R-HSA-76005 7/130 0.000444
Reactome_2022 Platelet Activation, Signaling And Aggregation R-HSA-76002 9/254 0.000444
Reactome_2022 Hemostasis R-HSA-109582 13/576 0.000444
Reactome_2022 Platelet Degranulation R-HSA-114608 7/125 0.000444
GO_Biological_Process_2021 ATF6-mediated unfolded protein response (GO:0036500) 3/9 0.005650
GO_Biological_Process_2021 regulation of insulin secretion (GO:0050796) 6/104 0.005650
Reactome_2022 Insertion Of Tail-Anchored Proteins Into Endoplasmic Reticulum Membrane R-HSA-9609523 3/22 0.012446
GO_Biological_Process_2021 positive regulation of peptide hormone secretion (GO:0090277) 4/43 0.017811
Reactome_2022 Insulin Processing R-HSA-264876 3/27 0.019352
[down] Significant terms: 72
Gene_set Term Overlap Adjusted P-value
Reactome_2022 Eukaryotic Translation Termination R-HSA-72764 18/90 7.313322e-22
Reactome_2022 Peptide Chain Elongation R-HSA-156902 18/86 7.313322e-22
Reactome_2022 Eukaryotic Translation Elongation R-HSA-156842 18/90 7.313322e-22
Reactome_2022 Nonsense Mediated Decay (NMD) Independent Of Exon Junction Complex (EJC) R-HSA-975956 18/92 8.430640e-22
GO_Biological_Process_2021 cytoplasmic translation (GO:0002181) 18/93 8.202504e-21
Reactome_2022 Viral mRNA Translation R-HSA-192823 17/90 2.034285e-20
Reactome_2022 Selenocysteine Synthesis R-HSA-2408557 17/90 2.034285e-20
Reactome_2022 Nonsense Mediated Decay (NMD) Enhanced By Exon Junction Complex (EJC) R-HSA-975957 18/112 2.141716e-20
Reactome_2022 Cap-dependent Translation Initiation R-HSA-72737 18/116 3.653633e-20
Reactome_2022 Response Of EIF2AK4 (GCN2) To Amino Acid Deficiency R-HSA-9633012 17/98 5.775833e-20
============================================================
Pathway enrichment: Epsilon
Gene sets: ['KEGG_2019_Mouse', 'GO_Biological_Process_2021', 'Reactome_2022']
Up-regulated genes : 96
Down-regulated genes: 90
============================================================
[down] Significant terms: 96
Gene_set Term Overlap Adjusted P-value
GO_Biological_Process_2021 cytoplasmic translation (GO:0002181) 29/93 2.964622e-44
GO_Biological_Process_2021 SRP-dependent cotranslational protein targeting to membrane (GO:0006614) 28/90 7.196346e-43
GO_Biological_Process_2021 cotranslational protein targeting to membrane (GO:0006613) 28/94 2.005921e-42
GO_Biological_Process_2021 nuclear-transcribed mRNA catabolic process, nonsense-mediated decay (GO:0000184) 29/113 5.161094e-42
GO_Biological_Process_2021 protein targeting to ER (GO:0045047) 28/103 2.341353e-41
Reactome_2022 Peptide Chain Elongation R-HSA-156902 27/86 4.047170e-41
Reactome_2022 Eukaryotic Translation Elongation R-HSA-156842 27/90 5.717472e-41
Reactome_2022 Eukaryotic Translation Termination R-HSA-72764 27/90 5.717472e-41
Reactome_2022 Nonsense Mediated Decay (NMD) Independent Of Exon Junction Complex (EJC) R-HSA-975956 27/92 8.577796e-41
Reactome_2022 Viral mRNA Translation R-HSA-192823 26/90 3.743559e-39
| condition | fate | direction | Gene_set | Term | Overlap | Adjusted P-value | |
|---|---|---|---|---|---|---|---|
| 0 | high_velocity | Alpha | up | KEGG_2019_Mouse | Protein processing in endoplasmic reticulum | 14/163 | 3.822697e-06 |
| 1 | high_velocity | Alpha | up | KEGG_2019_Mouse | Thyroid hormone synthesis | 9/73 | 3.211278e-05 |
| 2 | high_velocity | Alpha | up | KEGG_2019_Mouse | Circadian entrainment | 8/99 | 1.973558e-03 |
| 3 | high_velocity | Beta | up | KEGG_2019_Mouse | Protein processing in endoplasmic reticulum | 22/163 | 4.044027e-11 |
| 4 | high_velocity | Beta | up | GO_Biological_Process_2021 | IRE1-mediated unfolded protein response (GO:00... | 12/53 | 2.514449e-07 |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 25 | low_velocity | Beta | down | Reactome_2022 | Peptide Chain Elongation R-HSA-156902 | 18/86 | 7.313322e-22 |
| 26 | low_velocity | Beta | down | Reactome_2022 | Eukaryotic Translation Elongation R-HSA-156842 | 18/90 | 7.313322e-22 |
| 27 | low_velocity | Epsilon | down | GO_Biological_Process_2021 | cytoplasmic translation (GO:0002181) | 29/93 | 2.964622e-44 |
| 28 | low_velocity | Epsilon | down | GO_Biological_Process_2021 | SRP-dependent cotranslational protein targetin... | 28/90 | 7.196346e-43 |
| 29 | low_velocity | Epsilon | down | GO_Biological_Process_2021 | cotranslational protein targeting to membrane ... | 28/94 | 2.005921e-42 |
30 rows × 7 columns
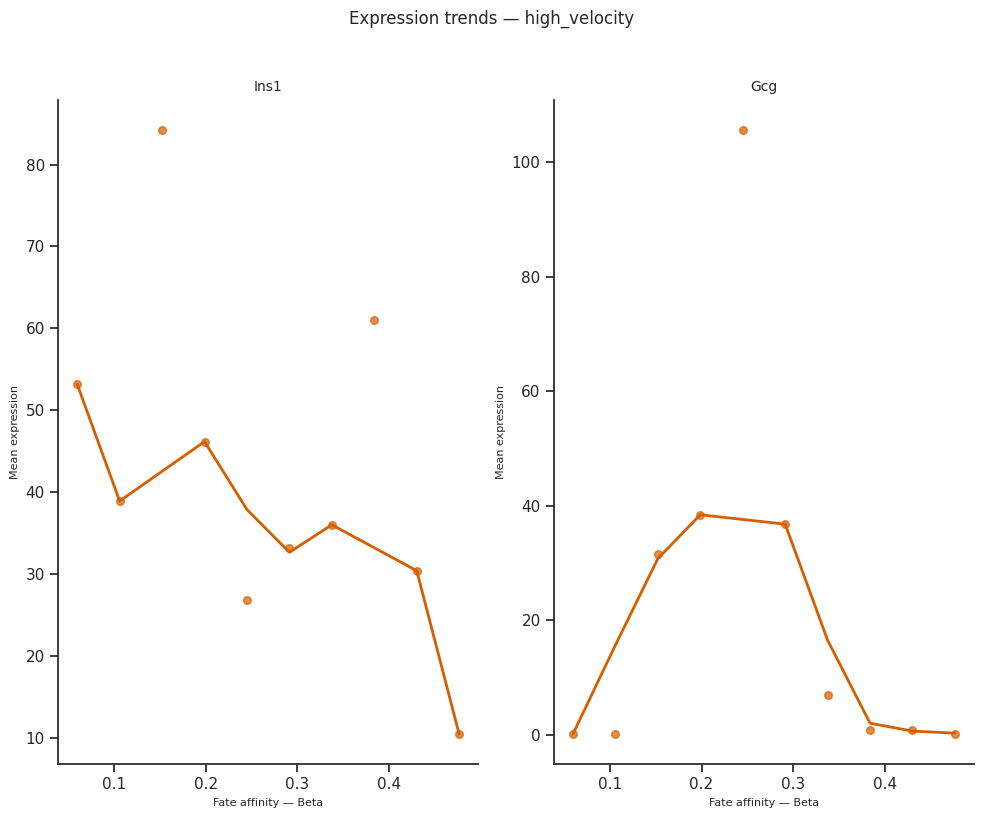
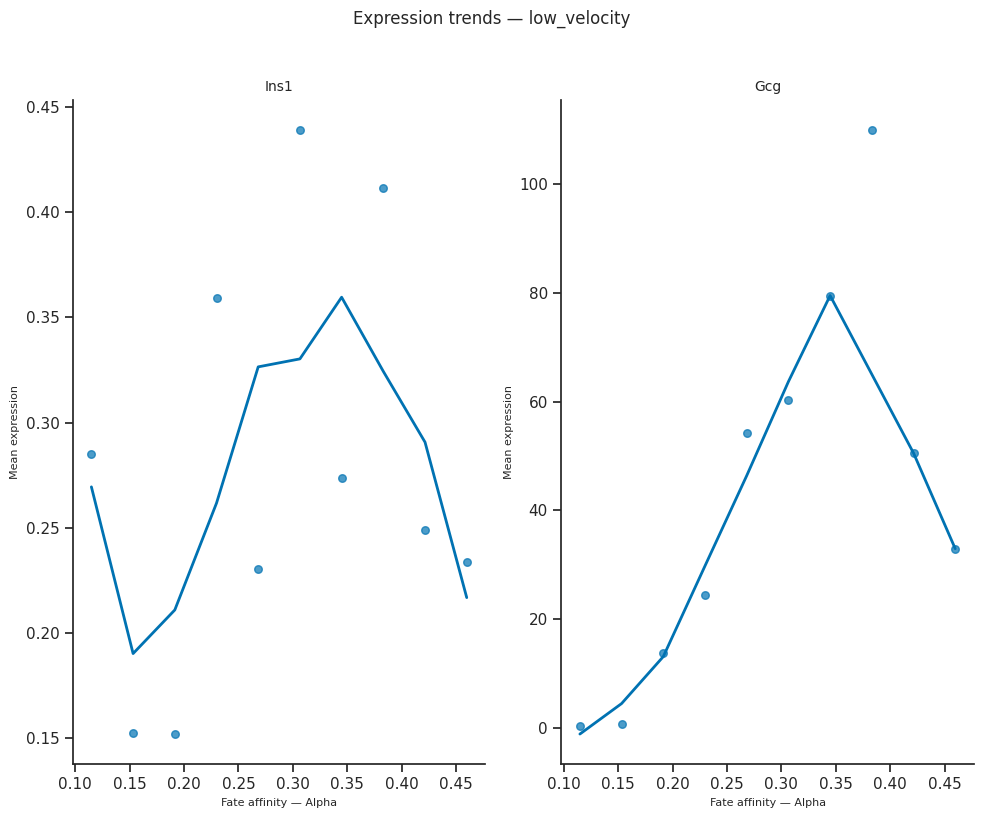
8. Expression trends along fate arms — per condition¶
plot_expression_trends orders cells by per-cell fate affinity (or pseudotime / radial distance) and plots smoothed expression of each gene.
Run it per condition side by side to see whether a marker’s expression trajectory differs between conditions — e.g. earlier onset, higher peak, delayed shutdown.
[31]:
# Expression trends, two markers, side-by-side per condition
from scCS.plot import plot_expression_trends
genes_of_interest = ["Ins1", "Gcg"] # Beta and Alpha markers
genes_of_interest = [g for g in genes_of_interest if g in adata_sub_shared.var_names]
assert genes_of_interest, "None of the requested markers are in the pancreas var_names"
for cond in mscorer.conditions:
mask = (adata_sub_shared.obs[mscorer.condition_obs_key].astype(str) == cond).values
sub = adata_sub_shared[mask].copy()
try:
fig = plot_expression_trends(
sub, results[cond],
genes=genes_of_interest,
x_axis="affinity",
figsize=(10, 4 * len(genes_of_interest)),
)
fig.suptitle(f"Expression trends — {cond}", y=1.02, fontsize=12)
plt.tight_layout()
plt.show()
except Exception as e:
print(f"plot_expression_trends failed for '{cond}': {e}")
9. Transfer labels to full adata¶
transfer_labels() writes per-cell scores from the shared embedding back to the full adata. This is identical to the single-condition workflow.
[32]:
# Transfer per-cell scores to full adata for all conditions
# PairScorer.transfer_labels writes condition-specific columns:
# e.g., cs_high_velocity_Alpha, cs_low_velocity_Alpha, etc.
mscorer.transfer_labels(results, prefix="cs_")
# Columns added to adata.obs
cs_cols = [c for c in adata.obs.columns if c.startswith("cs_")]
print("Columns added to adata.obs:", cs_cols[:8], "...")
# Visualize on UMAP
sc.pl.umap(adata, color=["clusters", "condition"], ncols=2, wspace=0.4)
[scCS] Labels transferred to adata.obs for 1876 / 3696 cells. Columns: ['cs_high_velocity_Alpha', 'cs_high_velocity_Beta', 'cs_high_velocity_Delta', 'cs_high_velocity_Epsilon', 'cs_high_velocity_dominant_fate', 'cs_high_velocity_entropy']
[scCS] Labels transferred to adata.obs for 1876 / 3696 cells. Columns: ['cs_low_velocity_Alpha', 'cs_low_velocity_Beta', 'cs_low_velocity_Delta', 'cs_low_velocity_Epsilon', 'cs_low_velocity_dominant_fate', 'cs_low_velocity_entropy']
Columns added to adata.obs: ['cs_high_velocity_Alpha', 'cs_high_velocity_Beta', 'cs_high_velocity_Delta', 'cs_high_velocity_Epsilon', 'cs_high_velocity_dominant_fate', 'cs_high_velocity_entropy', 'cs_high_velocity_nn_entropy', 'cs_low_velocity_Alpha'] ...
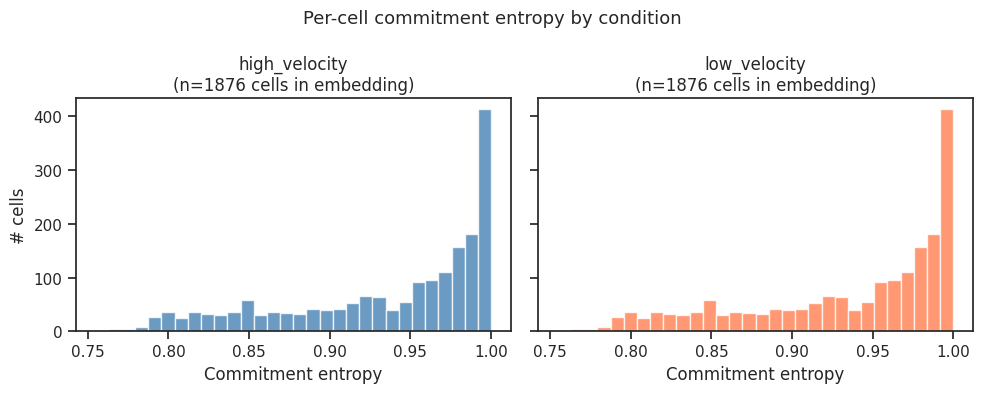
[33]:
# Compare entropy distributions between conditions
# After transfer_labels, columns are named cs_{condition}_{fate}
# For entropy, use the per-condition results directly from adata_sub
fig, axes = plt.subplots(1, 2, figsize=(10, 4), sharey=True)
colors = {"high_velocity": "steelblue", "low_velocity": "coral"}
for ax, cond in zip(axes, ["high_velocity", "low_velocity"]):
# Get entropy from the condition-specific result
res = results[cond]
if res.cell_scores is not None:
# Compute per-cell entropy from cell_scores
k = res.cell_scores.shape[1]
with np.errstate(divide="ignore", invalid="ignore"):
log_s = np.where(res.cell_scores > 0, np.log(res.cell_scores), 0.0)
ent = -np.sum(res.cell_scores * log_s, axis=1) / np.log(k)
ax.hist(ent, bins=30, color=colors[cond], edgecolor="white", alpha=0.8)
ax.set_title(f"{cond}\n(n={len(ent)} cells in embedding)")
ax.set_xlabel("Commitment entropy")
print(f"{cond}: mean entropy = {ent.mean():.3f} ± {ent.std():.3f}")
else:
ax.set_title(f"{cond}\n(no cell-level scores)")
axes[0].set_ylabel("# cells")
plt.suptitle("Per-cell commitment entropy by condition", fontsize=13)
plt.tight_layout()
plt.show()
high_velocity: mean entropy = 0.936 ± 0.063
low_velocity: mean entropy = 0.936 ± 0.063
Summary — scCS pairwise workflow¶
import scCS
# 1. Add condition column to adata.obs (or use existing one)
adata.obs["condition"] = ... # e.g., 'control', 'treated'
adata.obs["sample"] = ... # biological replicates
# 2. Initialize PairScorer
mscorer = scCS.PairScorer(
adata,
root="Ductal",
branches=["Alpha", "Beta", "Delta", "Epsilon"],
condition_obs_key="condition",
obs_key="clusters",
)
# 3. Build SHARED embedding on pooled data
mscorer.build_embedding(ordering_metric="pseudotime")
mscorer.refit_pseudotime(scale_01=False) # preserve ordering
mscorer.fit()
# ── Tier 1: Score each condition ──────────────────────────────────────────────
results = mscorer.score_all_conditions(cell_level=True, n_bootstrap=200)
mscorer.plot_star_grid(results)
mscorer.plot_rose_grid(results) # per-condition rose plots
# ── Tier 2: Statistical comparison ───────────────────────────────────────────
delta = mscorer.compute_delta_CS("treated", "control", n_bootstrap=500)
stats = mscorer.compare_conditions(results, n_permutations=1000)
mscorer.plot_affinity_distributions(results, plot_type="violin")
# ── Tier 3: Trajectory-level analysis ────────────────────────────────────────
lmm = mscorer.fit_mixed_model(results, replicate_key="sample")
shift = mscorer.trajectory_shift(results, n_bootstrap=500)
mscorer.plot_trajectory_shift(shift)
# ── Downstream §6: Driver genes per condition ────────────────────────────────
# Loop conditions: mask adata_sub per condition, call get_velocity_drivers /
# get_deg_drivers, then collect into one (condition, fate, gene) table.
# ── Downstream §7: Pathway enrichment per condition ──────────────────────────
# Loop conditions: call run_enrichment_per_fate on the per-condition DEG
# drivers, collect into one (condition, fate, direction, Term) table.
# ── Downstream §8: Expression trends per condition ───────────────────────────
# Loop conditions: call plot_expression_trends on the masked adata_sub plus
# results[cond] for side-by-side per-condition trajectories.
# ── Transfer per-cell labels back to full adata for UMAP / downstream ────────
mscorer.transfer_labels(adata, results)
Interpretation guide¶
Result |
Interpretation |
|---|---|
ΔnCS > 0, CI excludes 0 |
Condition A has stronger commitment toward that fate pair |
Permutation p < 0.05 |
Per-cell fate affinities differ significantly between conditions |
LMM condition coefficient significant |
Condition effect on fate affinity, controlling for replicate |
KS p < 0.05 + large Wasserstein |
Cells in one condition are systematically further along the trajectory |
Top driver gene present in one condition only |
Condition-specific regulator candidate |
Pathway enriched in one condition only |
Differentially activated pathway |
Expression trend earlier/later in one condition |
Shifted developmental timing for that gene |
For single-condition analysis, see Single Condition Analysis.ipynb. For 3+ condition analysis, see Multiple Comparison Analysis.ipynb.